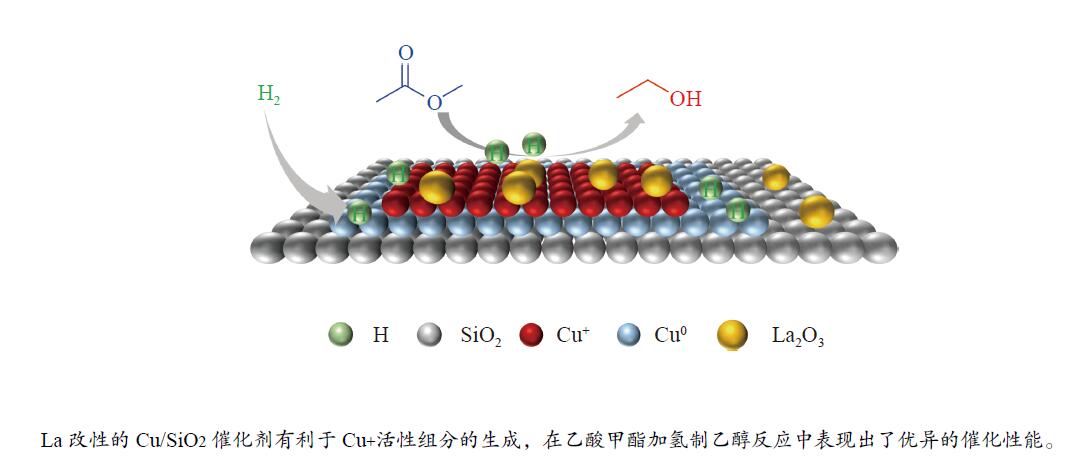
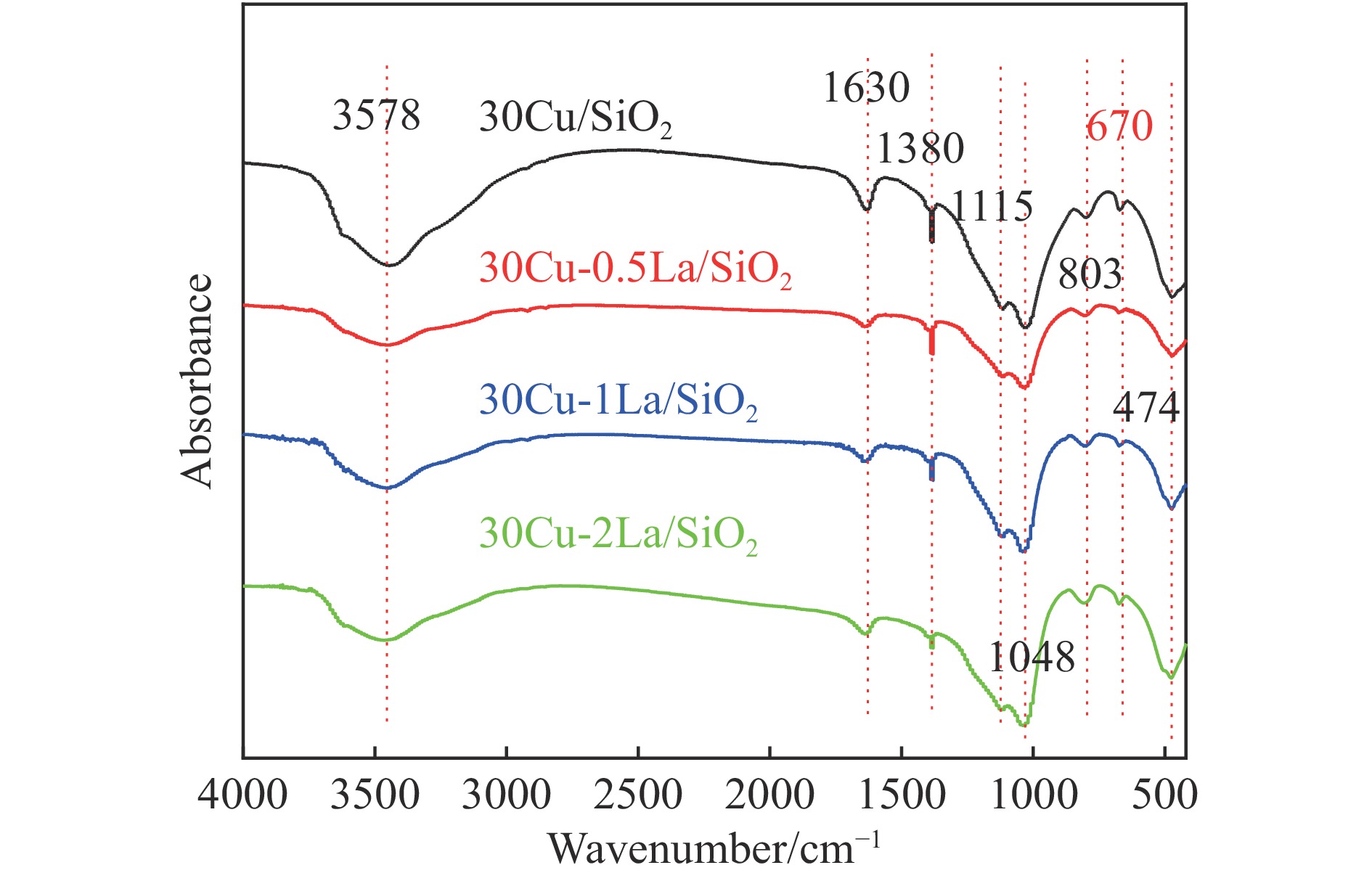
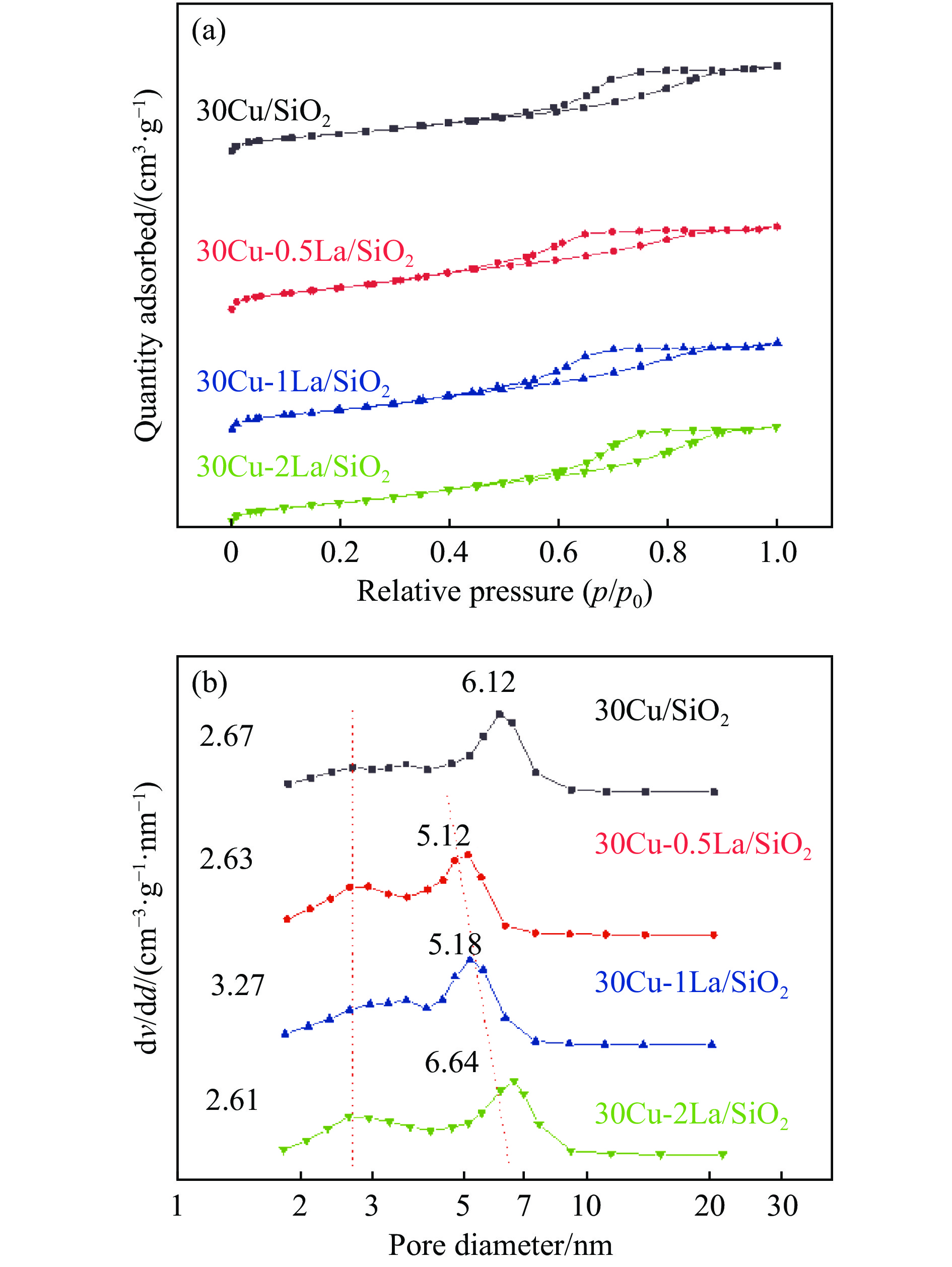
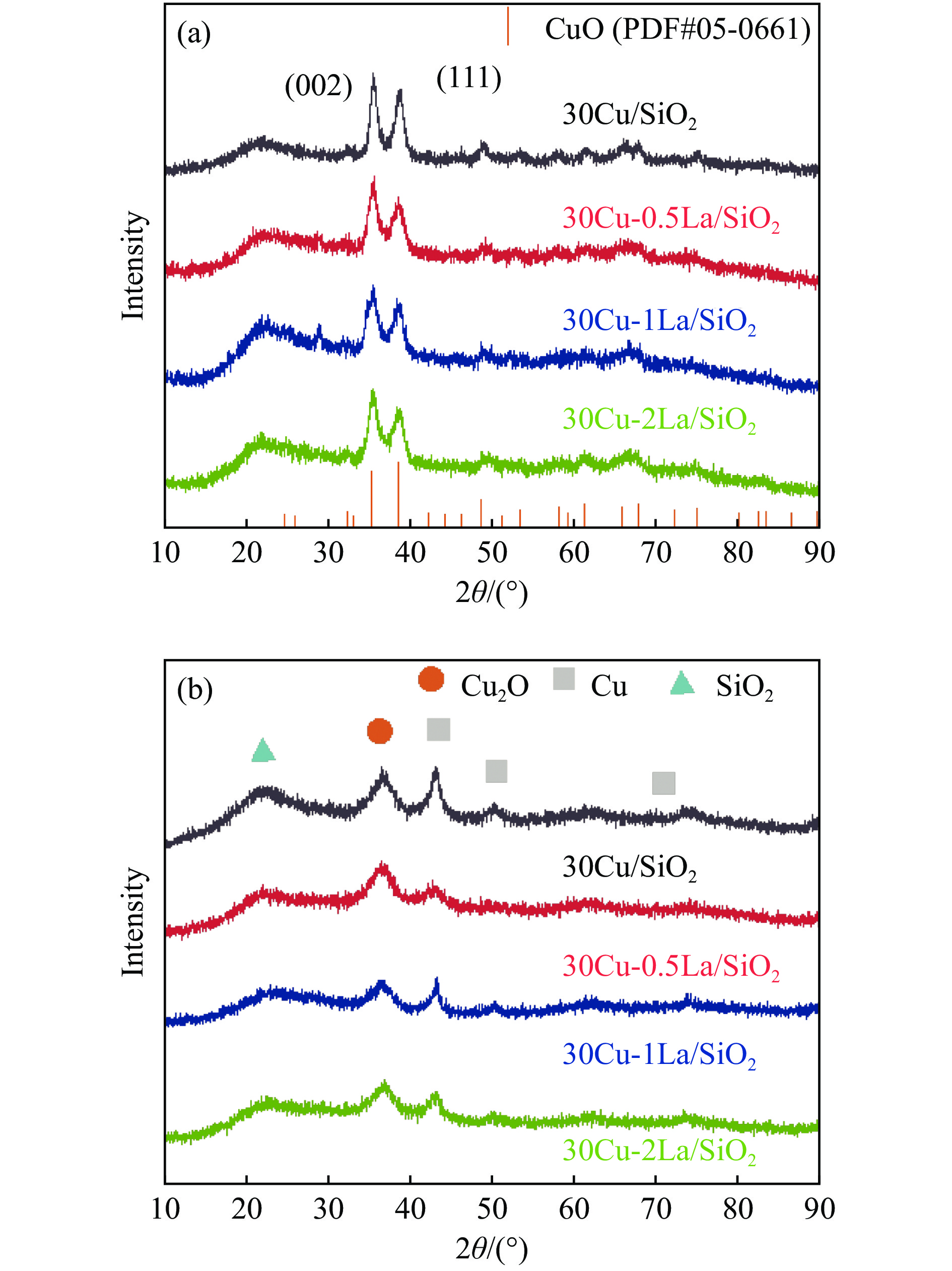
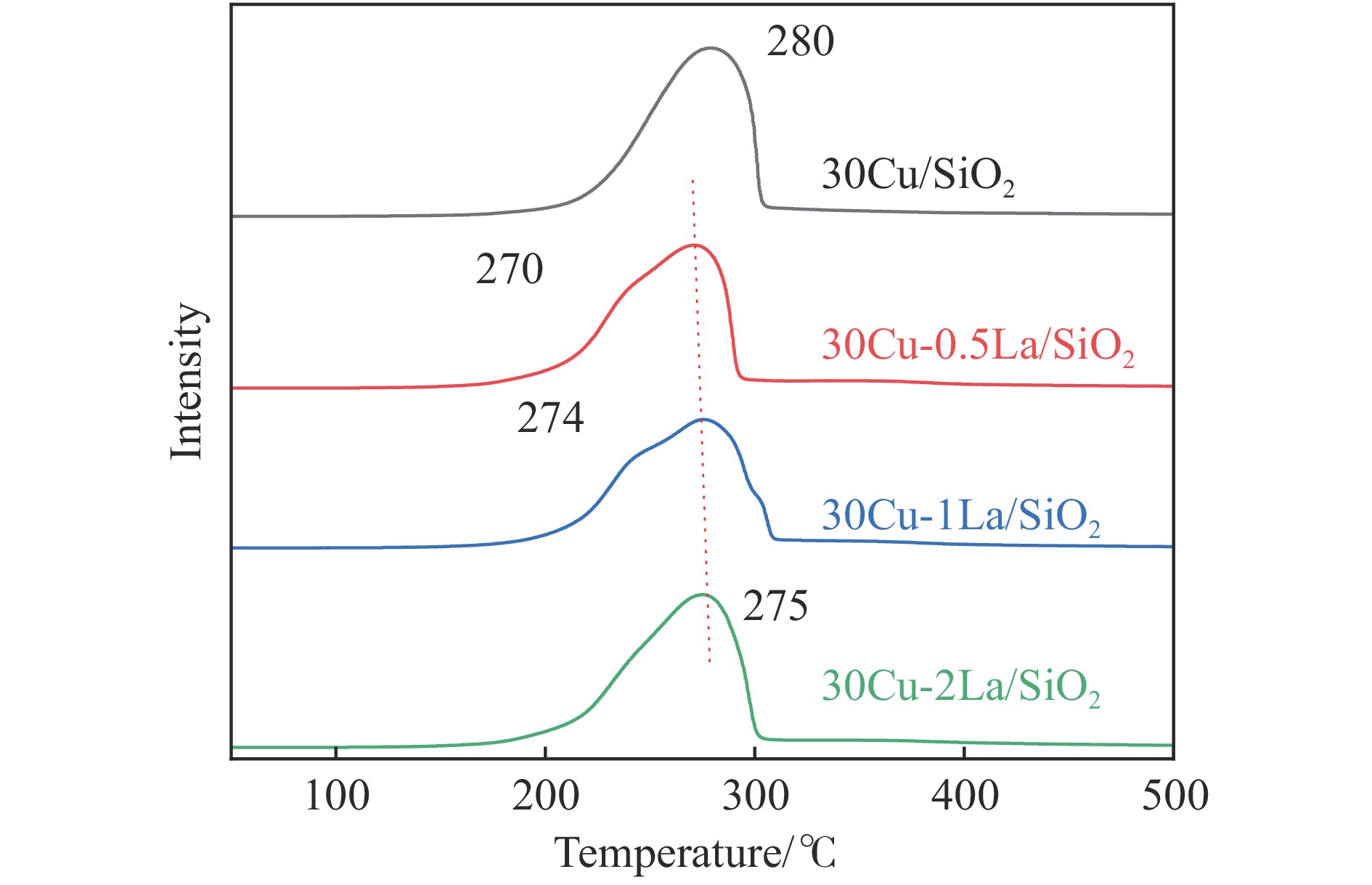
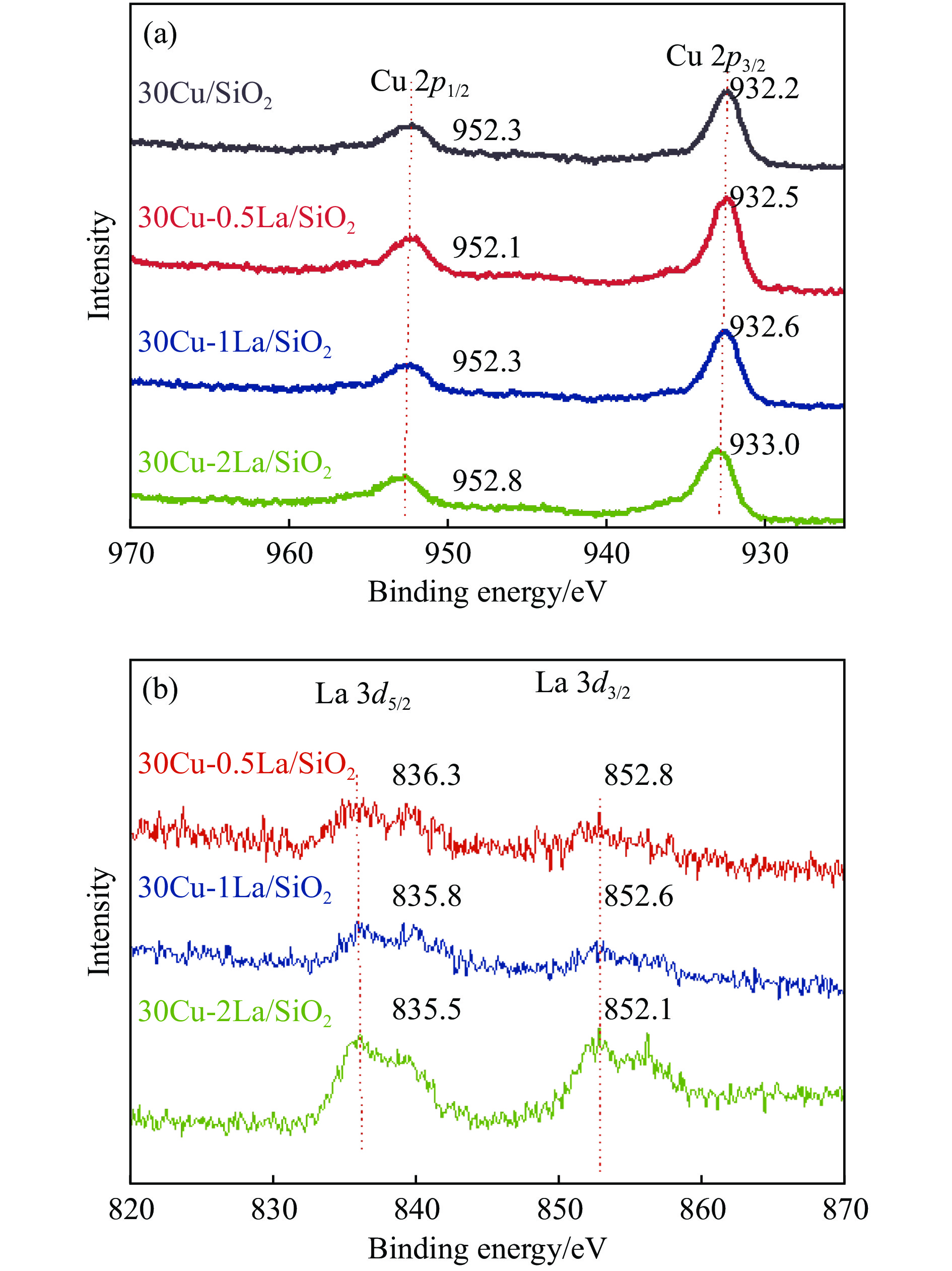
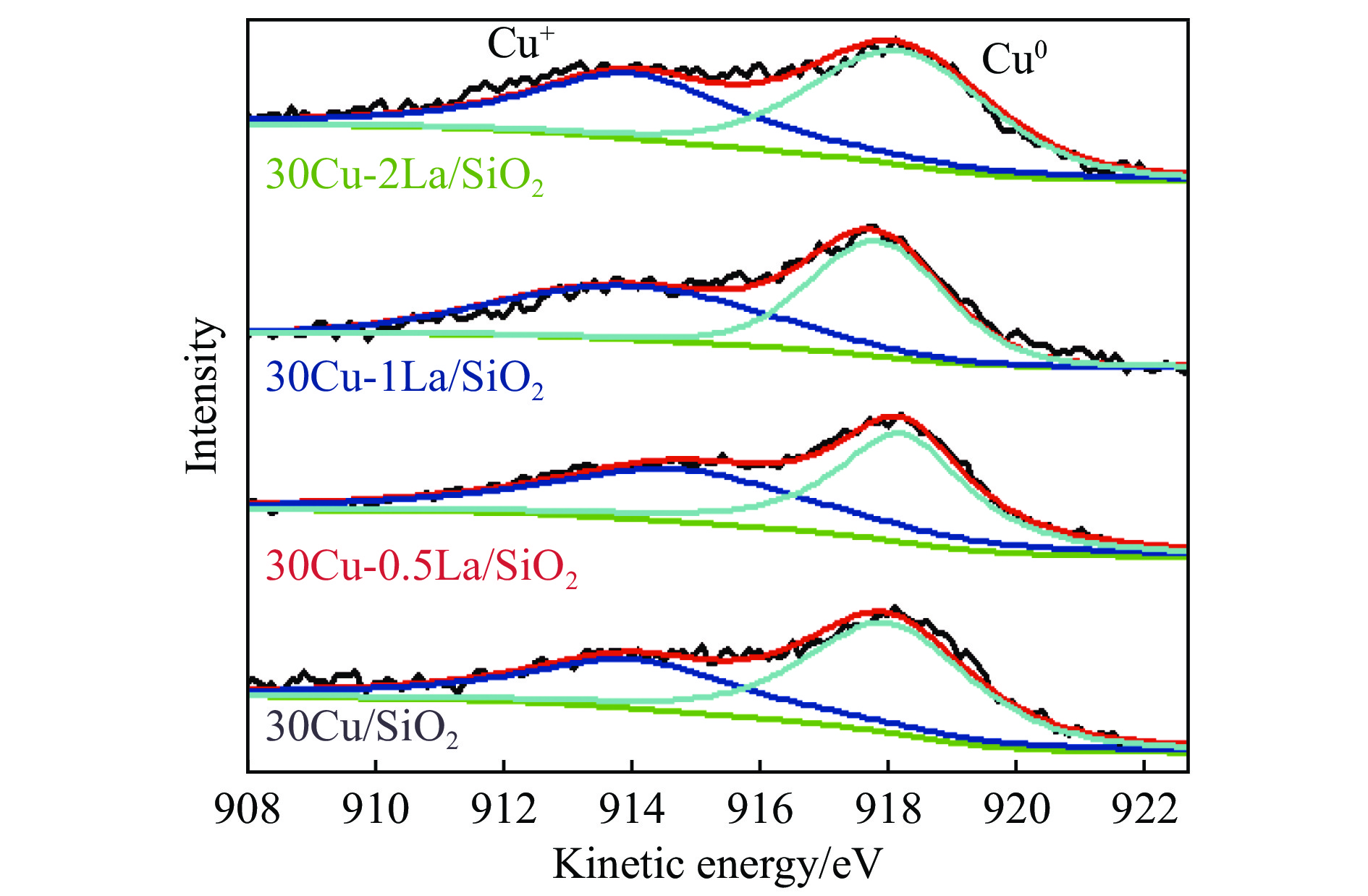
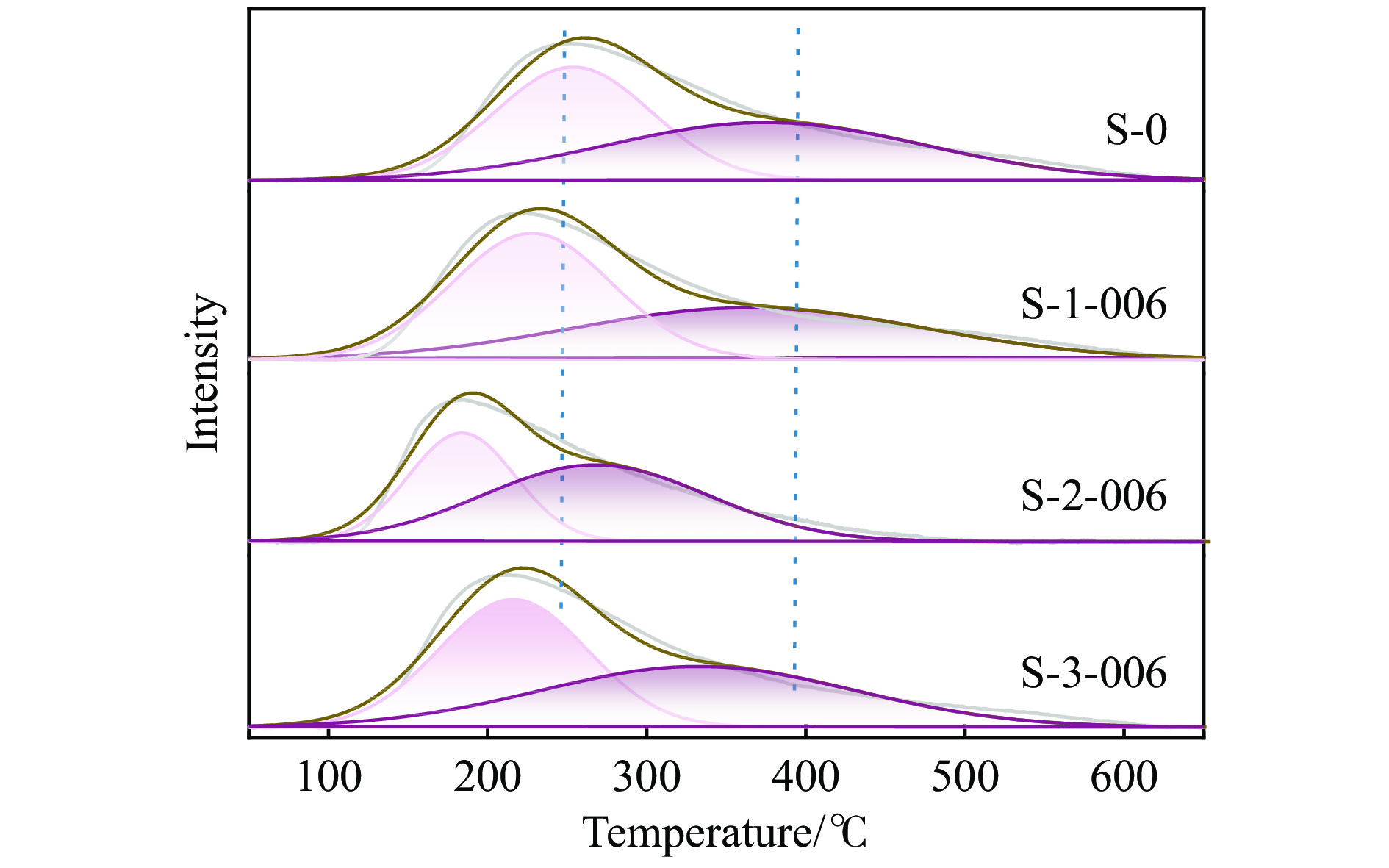
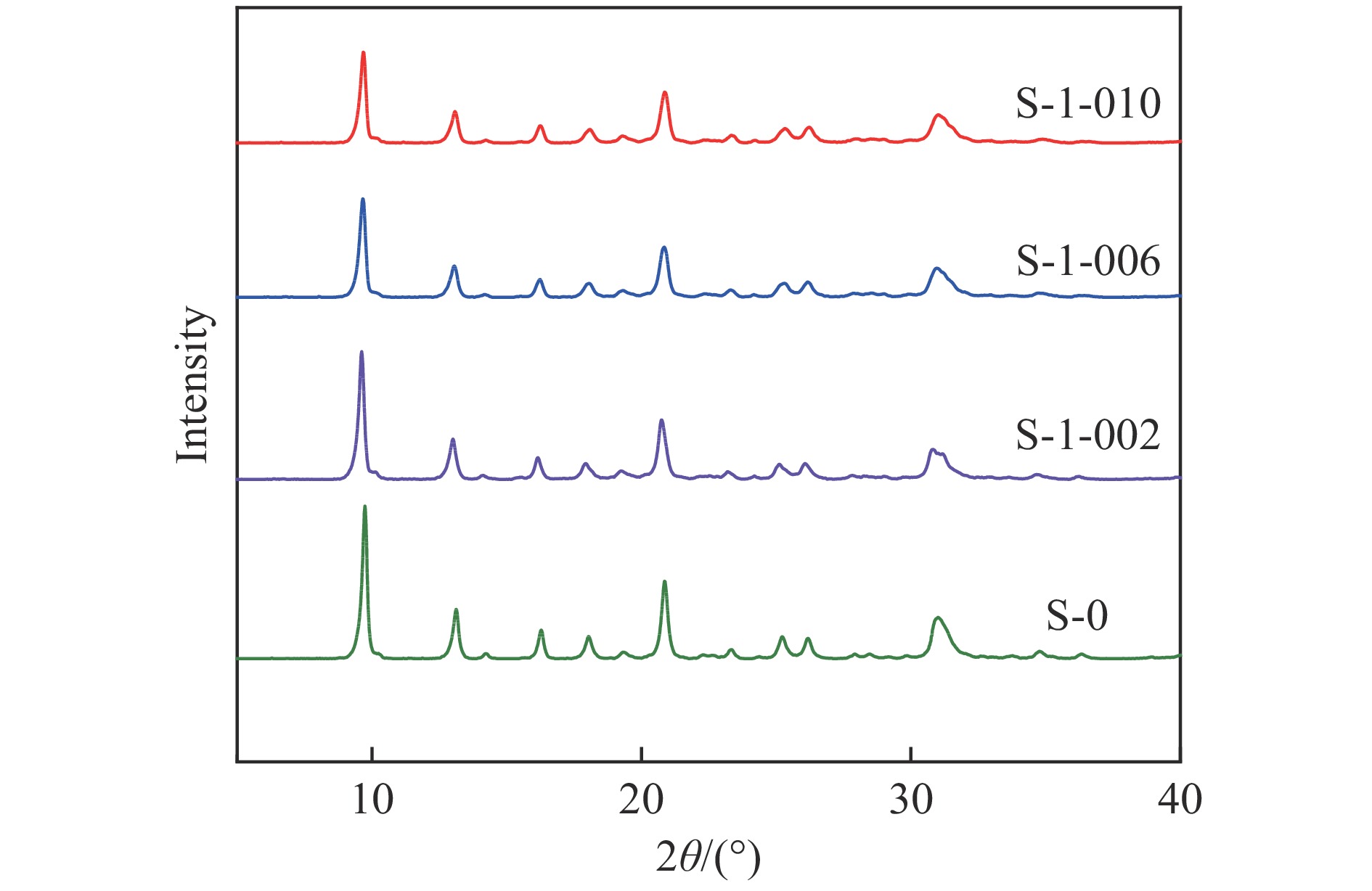
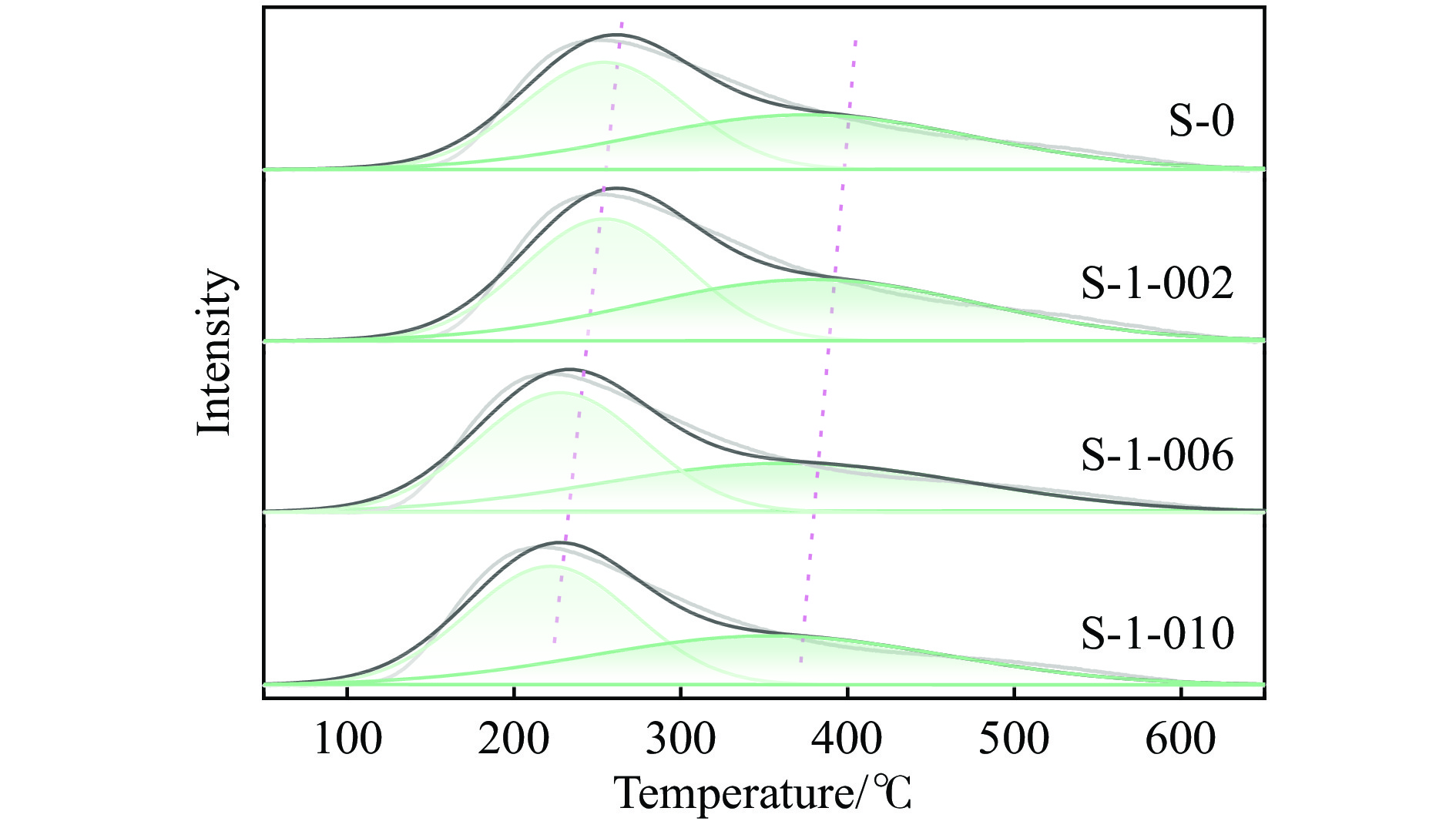
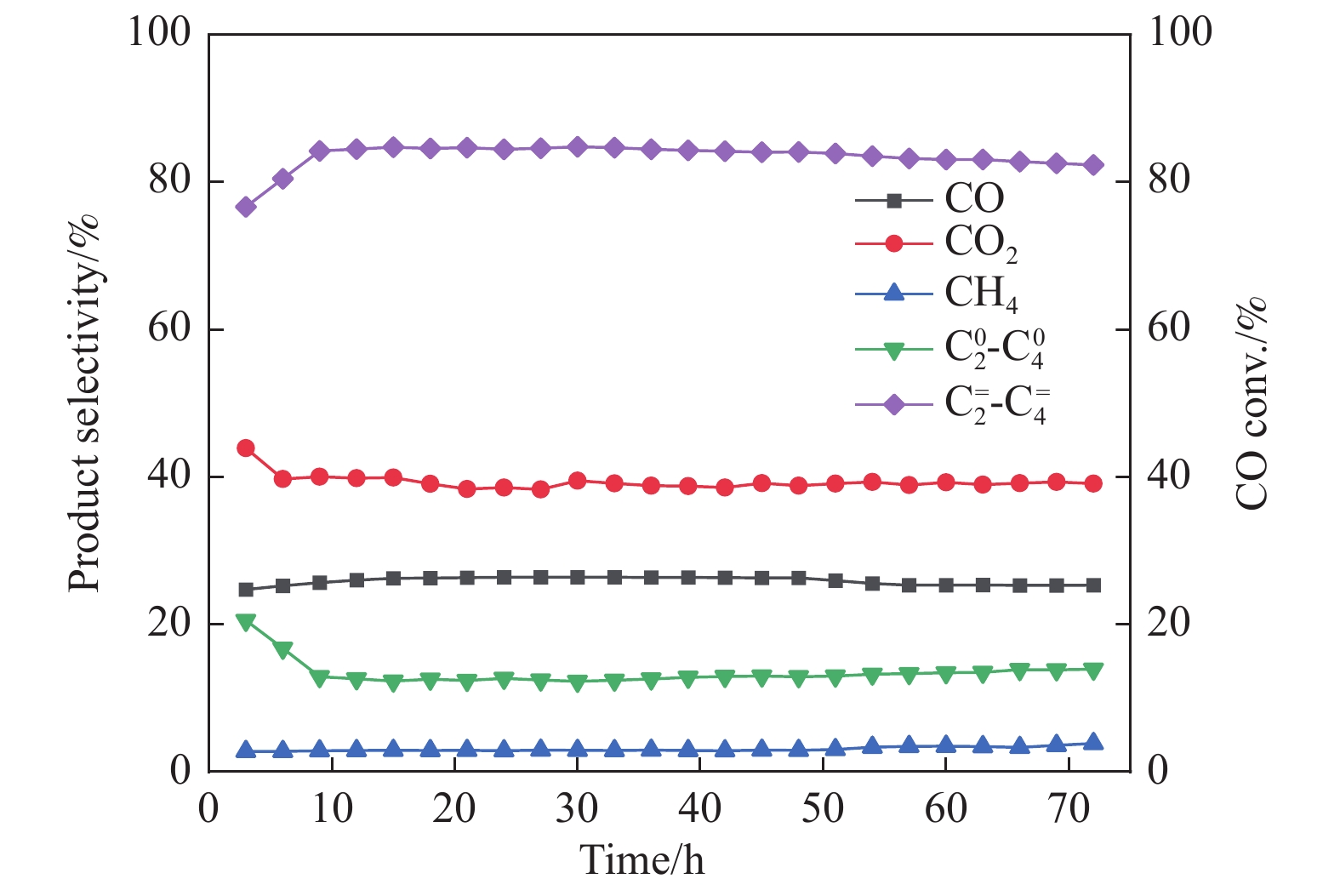
摘要: 采用蒸氨法制备了镧(La)改性的负载型铜硅(Cu/SiO2 )催化剂,并对其乙酸甲酯(MeOAc)气相加氢制乙醇(EtOH)的催化性能进行了研究。采用N2 吸附-脱附(N2 adsorption-desorption)、X射线粉末衍射(XRD)、电感耦合等离子体发射光谱(ICP-OES)、氢气程序升温还原(H2 -TPR)、傅里叶红外光谱(FT-IR)、高分辨透射电镜(HRTEM)、光电子能谱(XPS)和原子发射光谱仪(AES)等手段对催化剂进行了的表征,发现La物种的加入产生了较多的层状硅酸铜,增强了Cu和La物种之间的相互作用。La物种的加入在结构方面提高了催化剂的比表面积,降低了铜物种的粒径,提高了铜物种的分散度;在电子还原调控方面提高了Cu+ 的含量,增强了催化剂吸附酰基和甲氧基的能力。与未改性的Cu/SiO2 催化剂相比,镧改性后Cu/SiO2 催化剂的乙酸甲酯加氢性能得到大幅提升;其中La掺杂量0.5%的Cu/SiO2 催化剂表现出最佳的催化性能,乙酸甲酯转化率达98.5%,乙醇的总收率为97.0%。
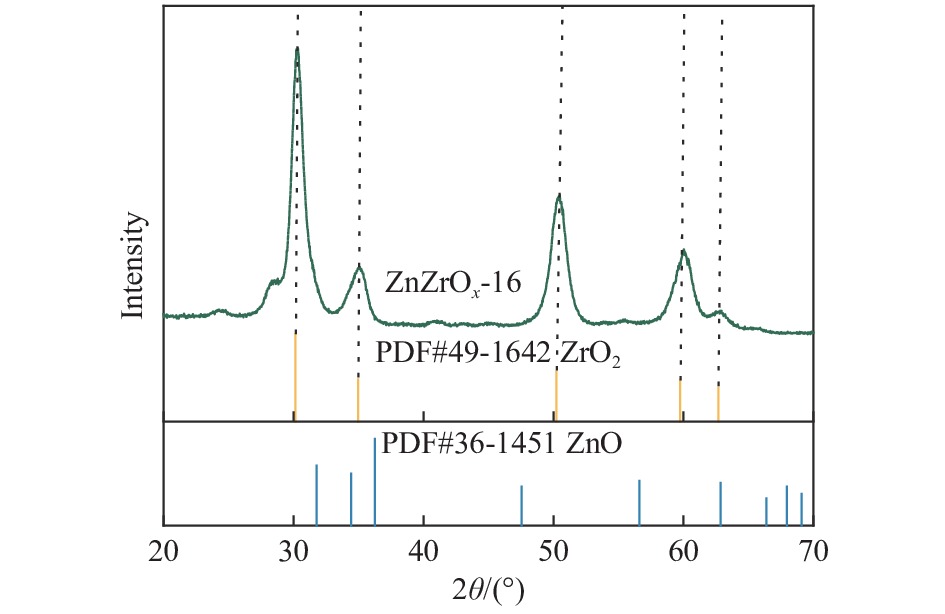
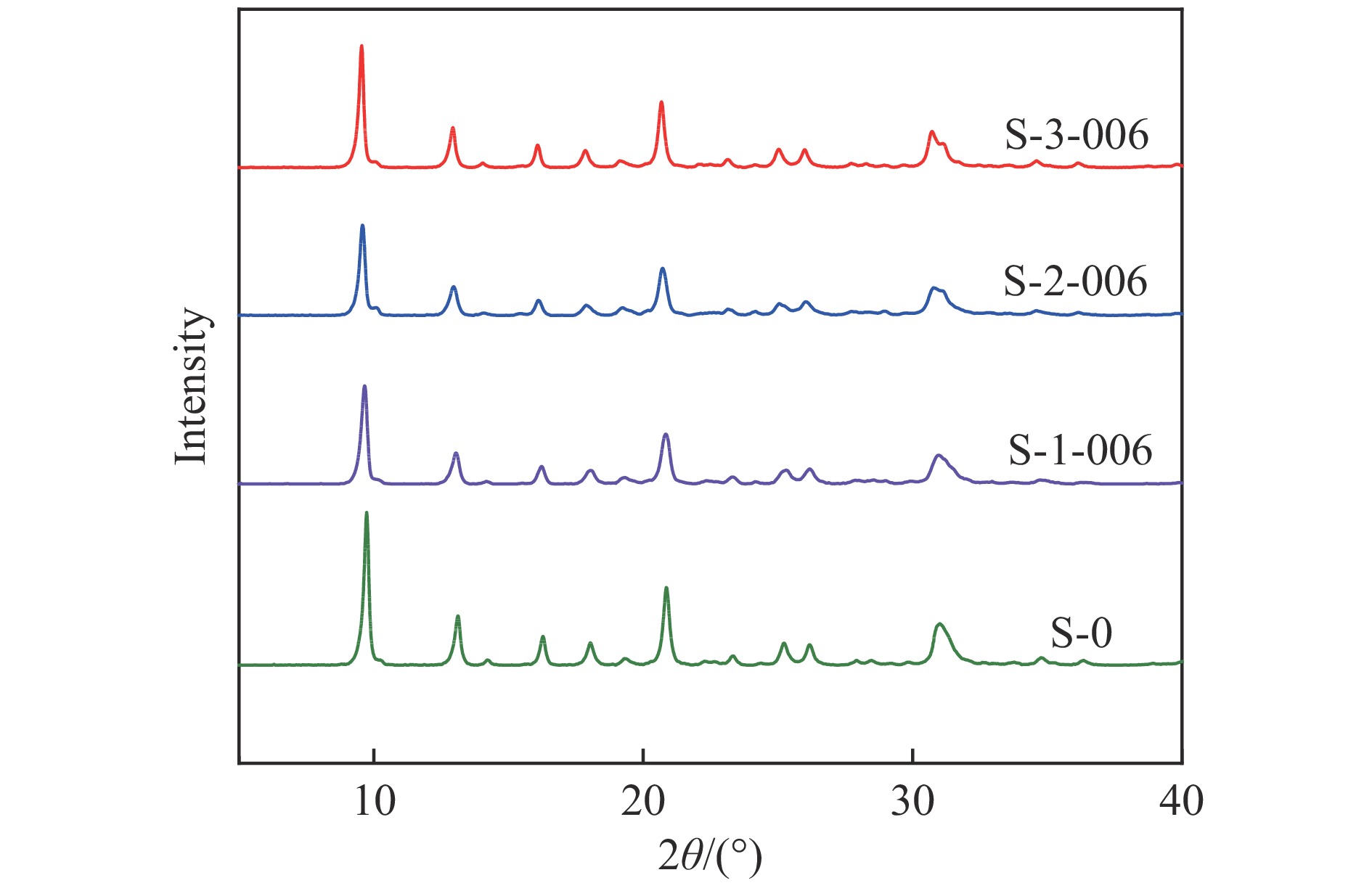
摘要: 采用ZnZrOx 2 吸附-脱附、NH3 -TPD对分子筛进行了表征,并考察了碱处理后催化剂的STO反应性能。结果表明,采用0.06 mol/L的三种有机碱后处理均可在SAPO-34分子筛表面刻蚀出部分多级孔道,从而在STO反应中加速金属氧化物表面形成的中间过渡物种从金属氧化物表面扩散进入SAPO-34分子筛孔道,提高了催化剂在STO反应中CO的转化率,同时,三种碱处理均可降低SAPO-34分子筛的酸量及酸强度,从而提高催化剂在STO反应中低碳烯烃选择性;采用0.02−0.10 mol/L的三乙胺处理SAPO-34分子筛,均在SAPO-34分子筛表面刻蚀出的多级孔,提高了STO反应中CO的转化率,且0.02和0.06 mol/L的三乙胺溶液处理后的SAPO-34分子筛,酸强度和酸量的降低,抑制了甲烷的形成和烯烃的加氢,因此,随着碱处理浓度从0、0.02到0.06 mol/L逐步提高,催化剂对低碳烯烃的选择性逐步提高。其中,在400 ℃,3.0 MPa和GHSV=3600 mL/(g∙h)条件下,采用0.06 mol/L的三乙胺处理的SAPO-34物理混合ZnZrOx
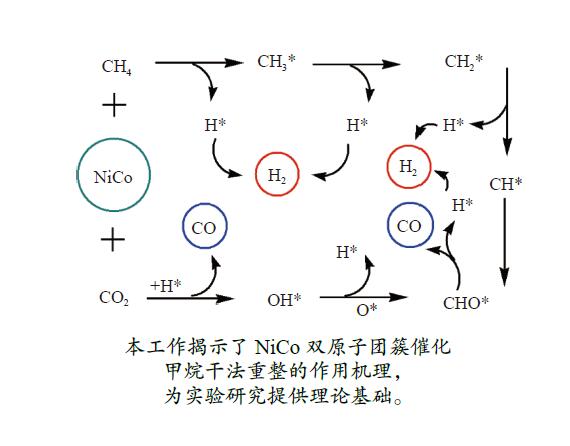
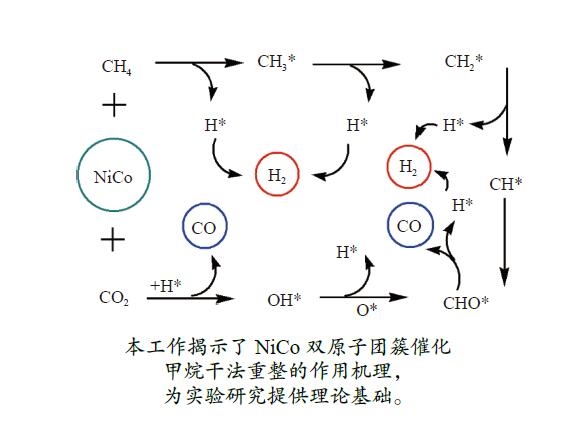
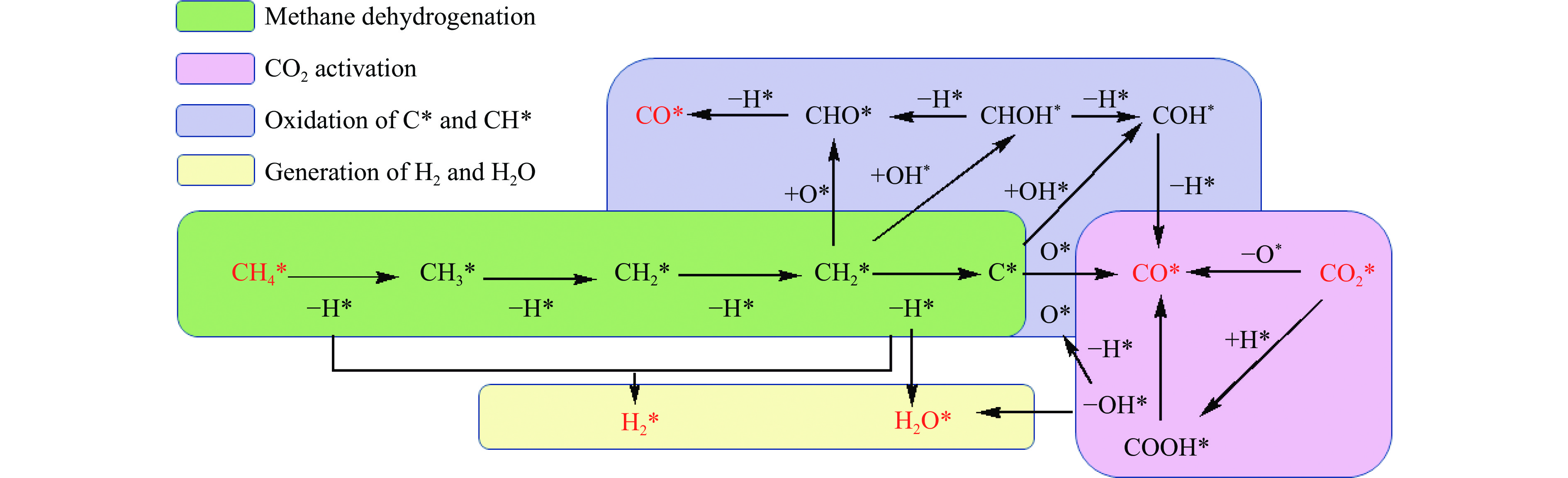
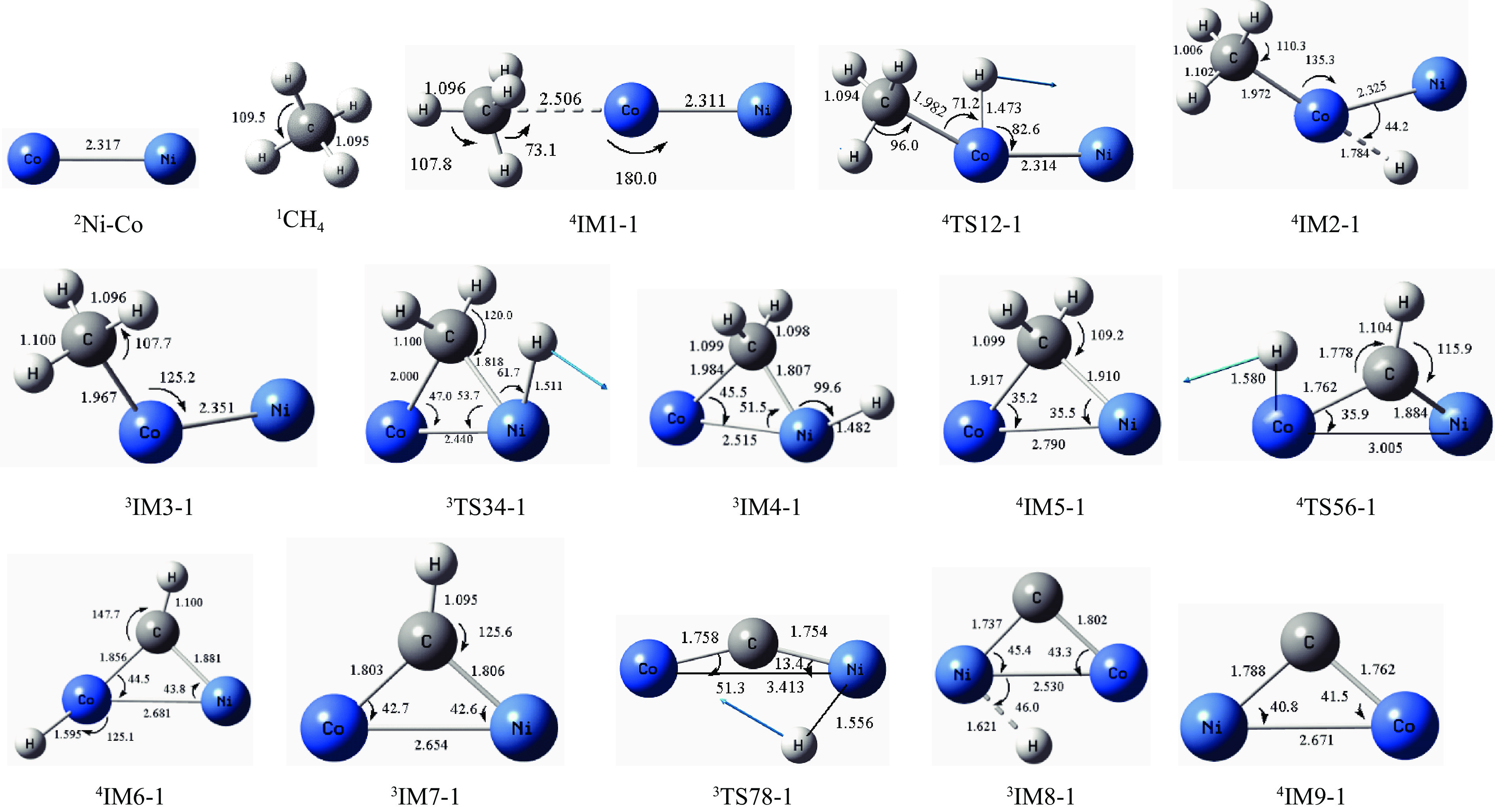
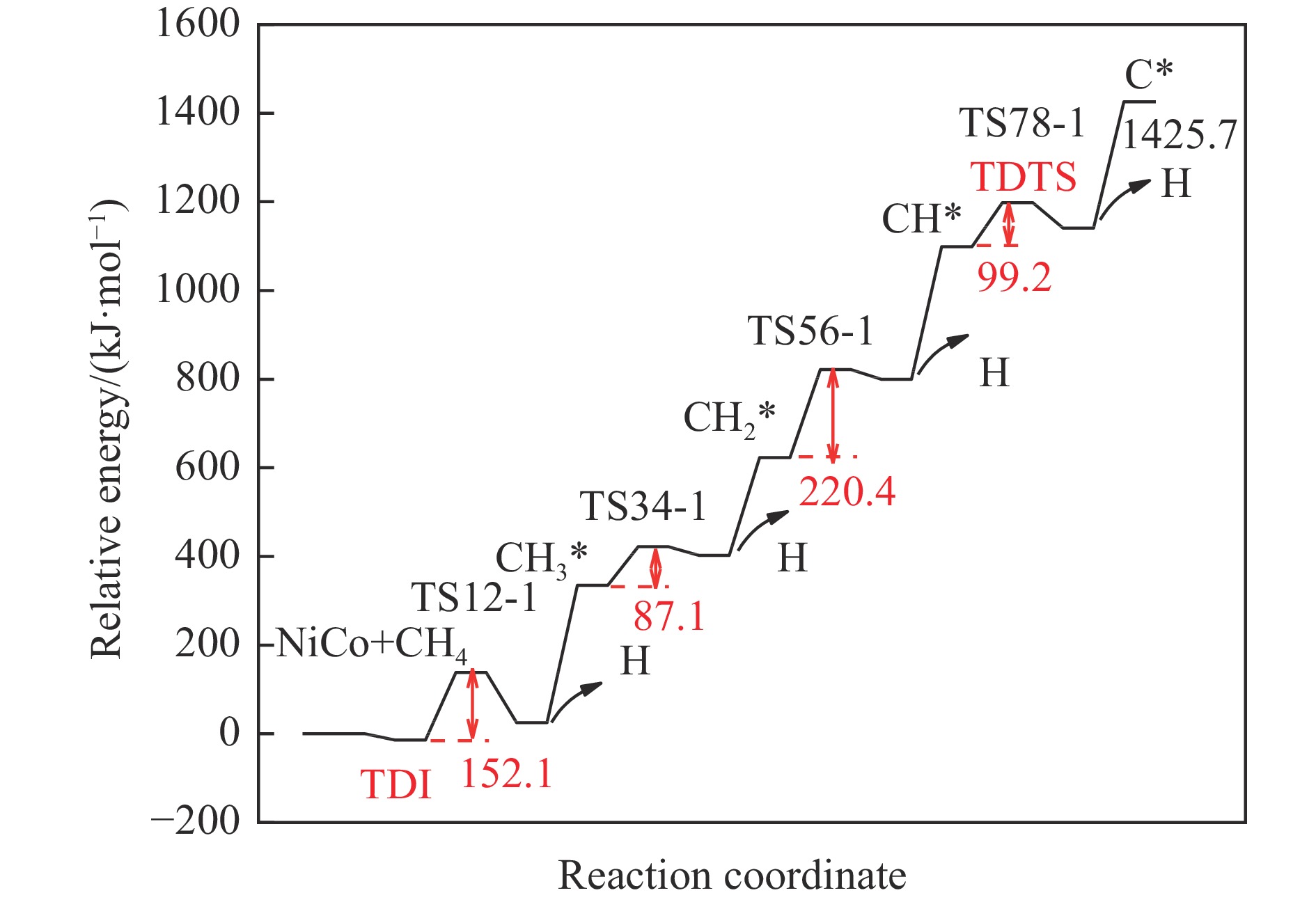
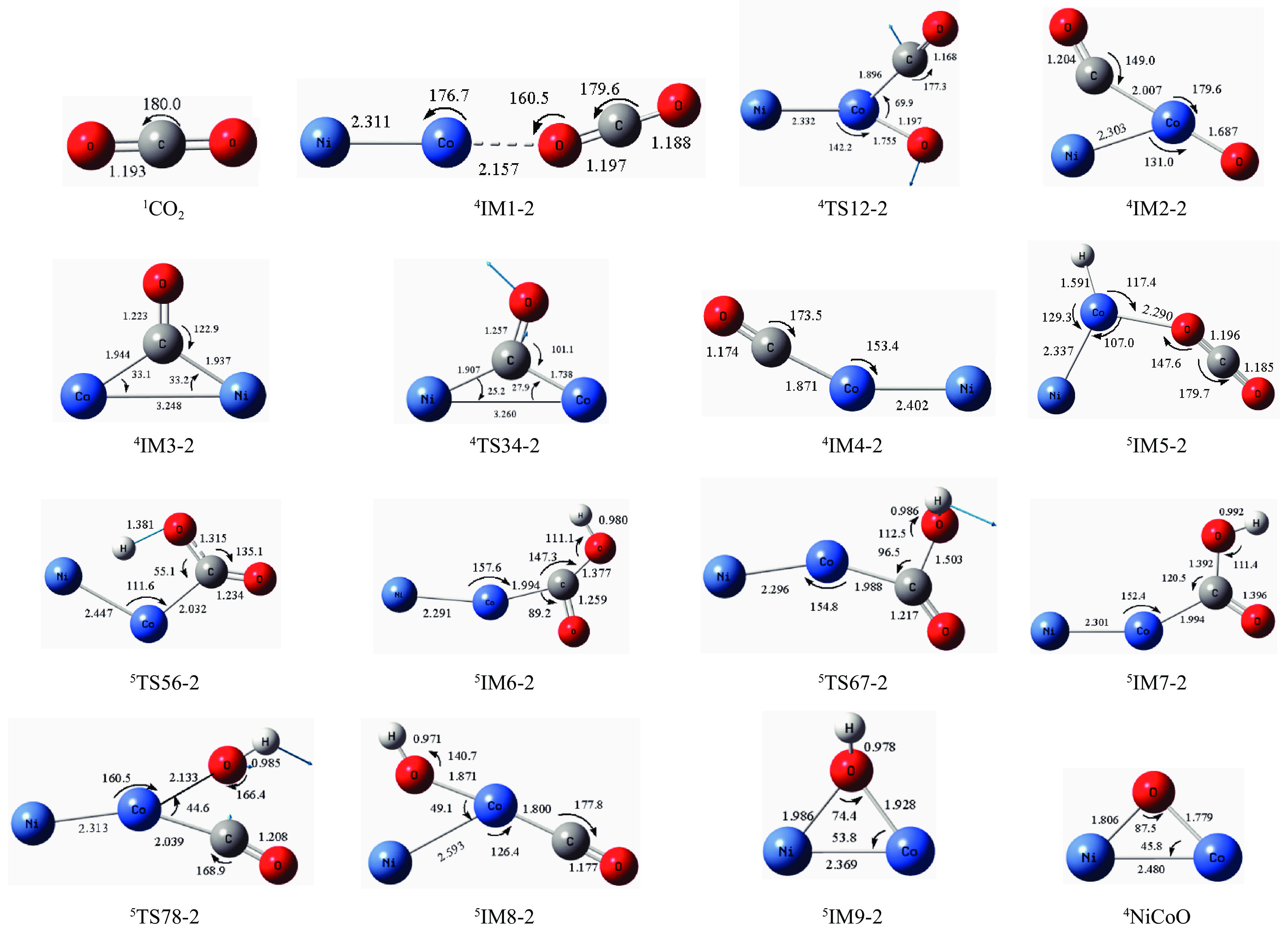
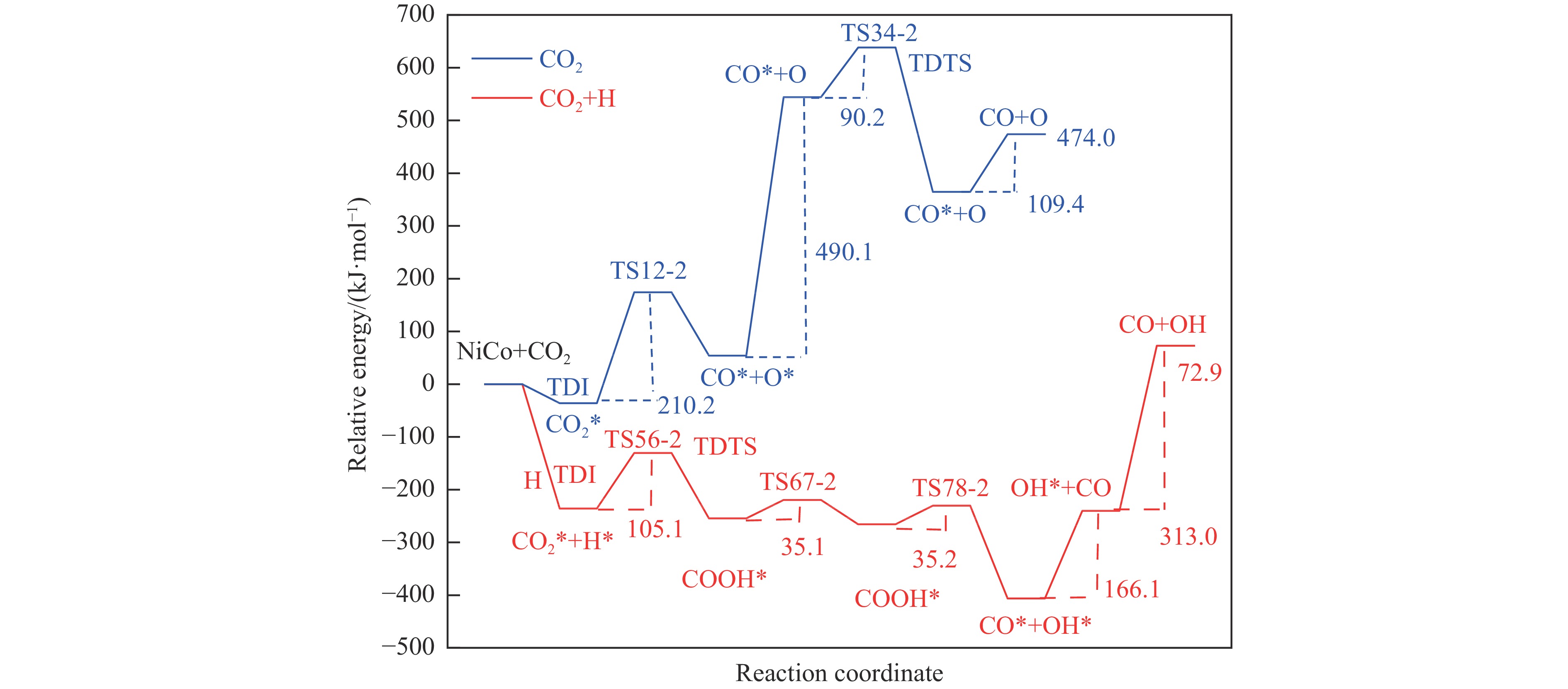
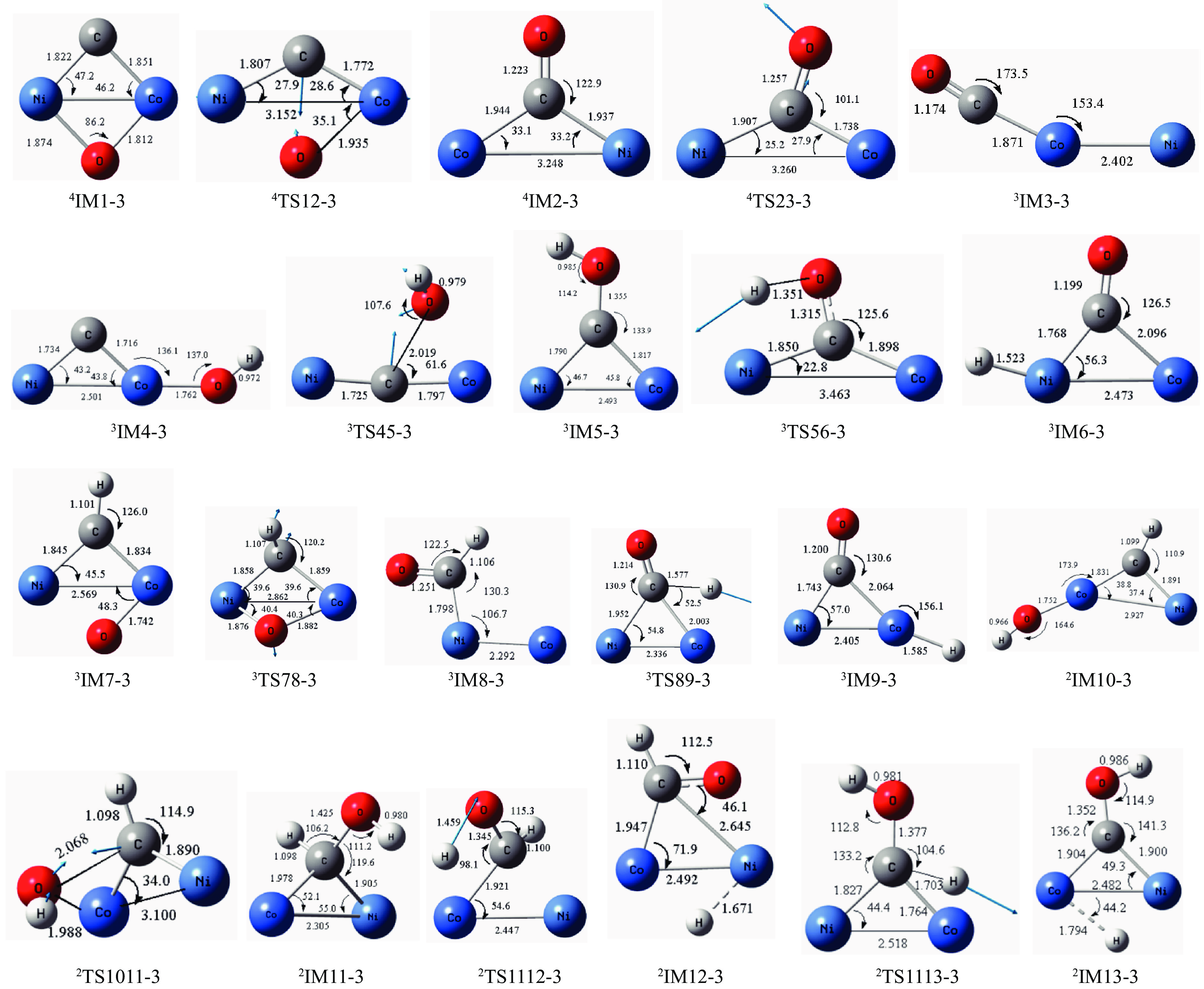
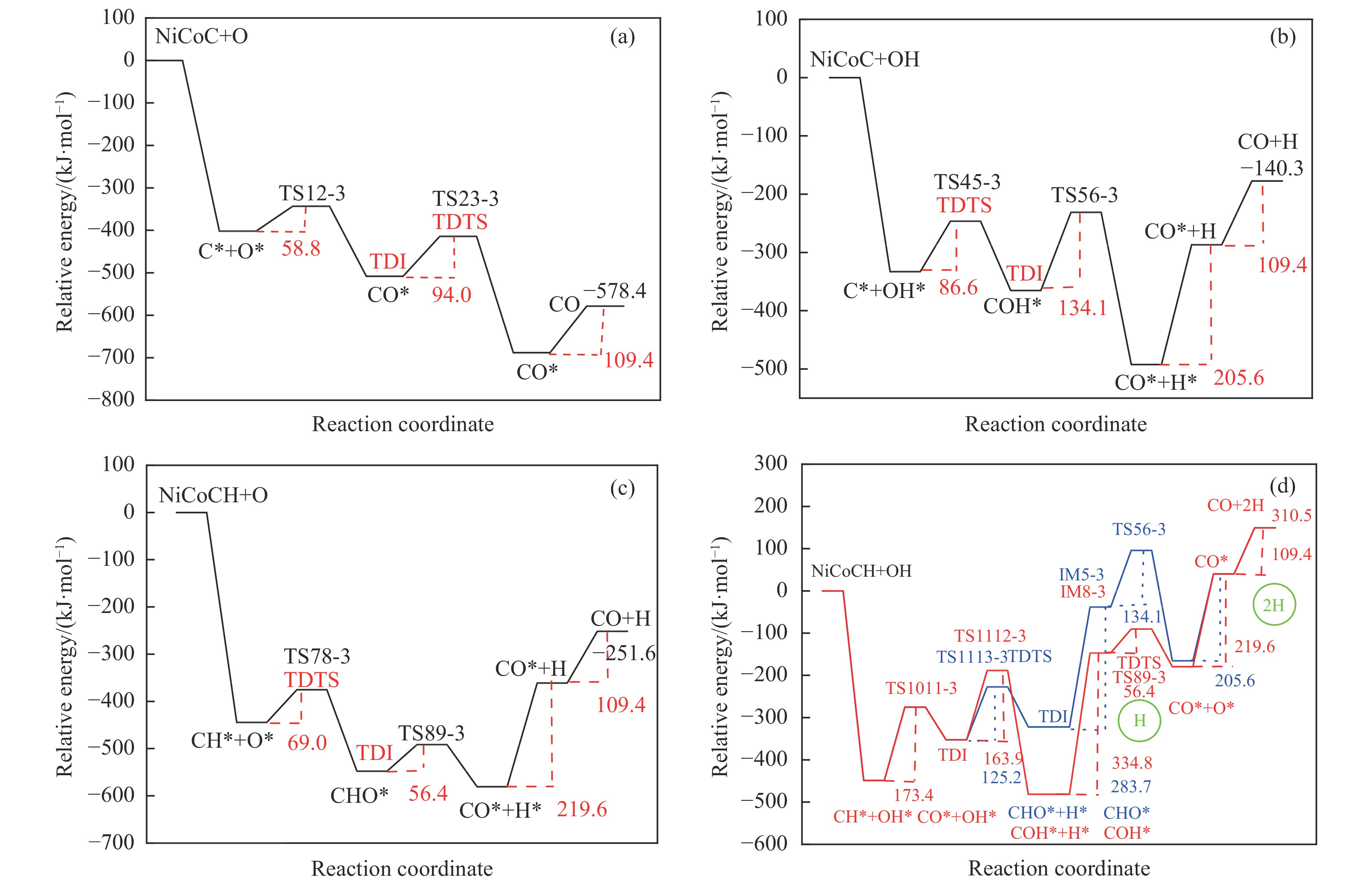
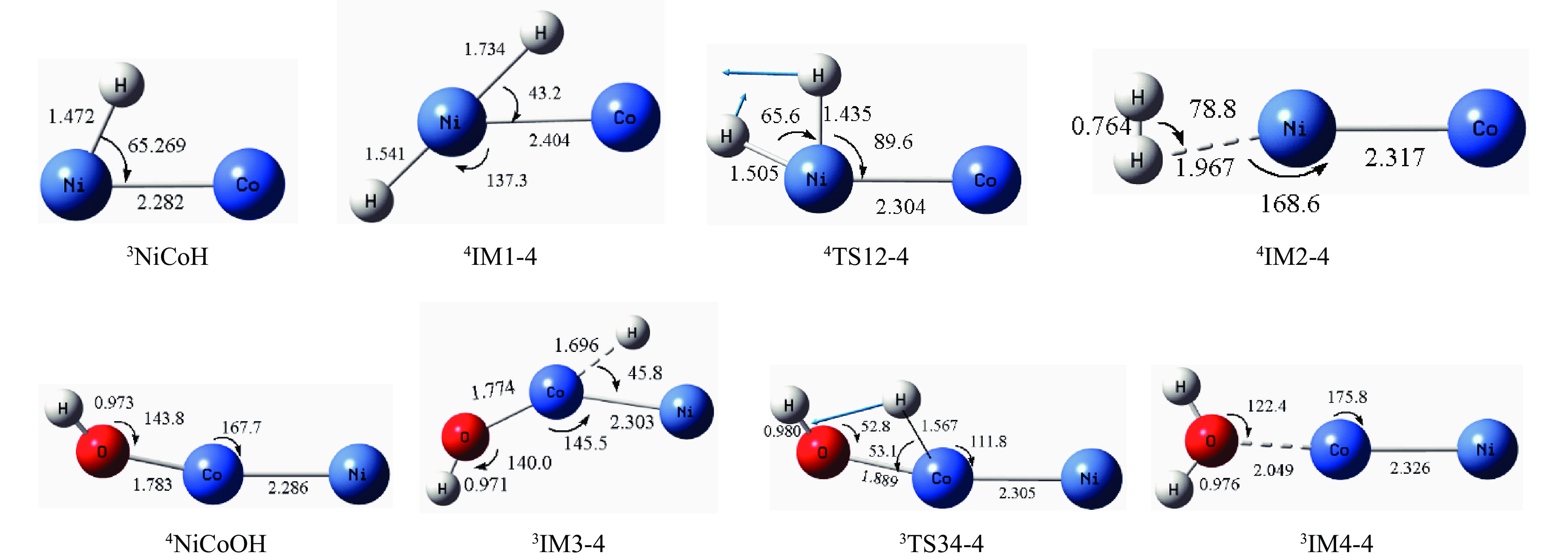
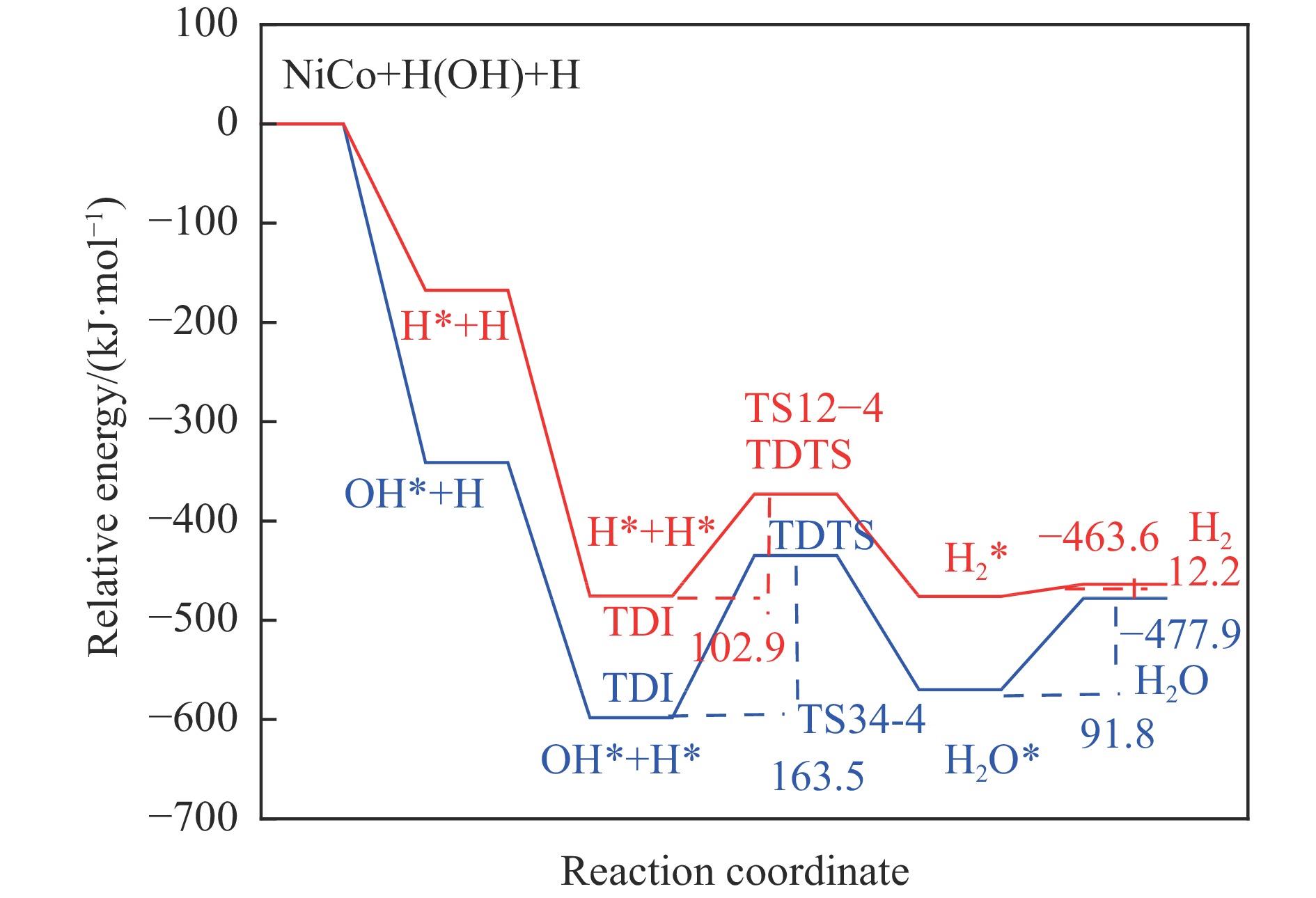
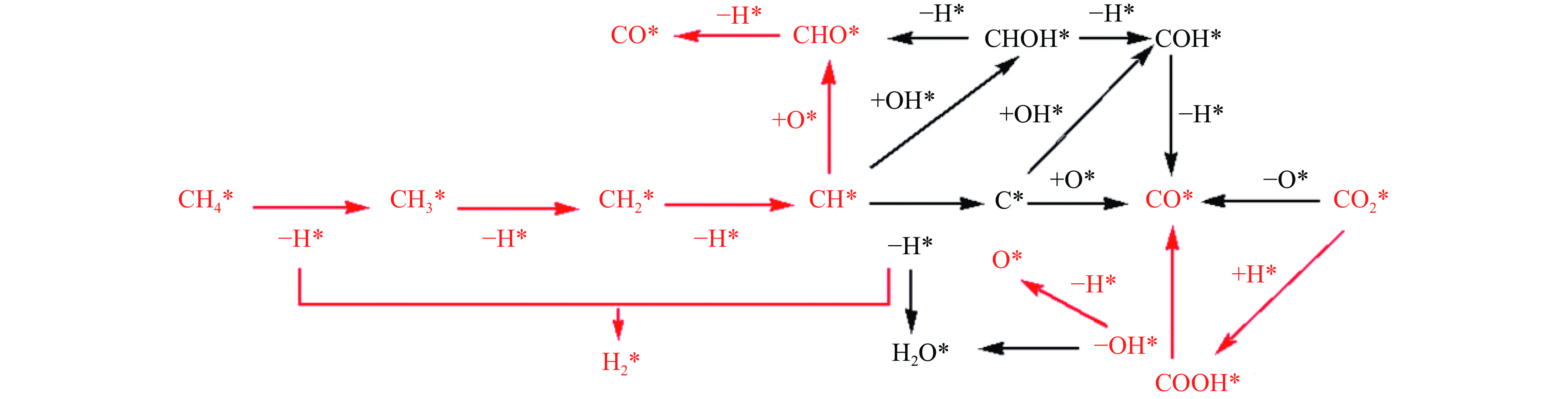
摘要: 本研究采用密度泛函理论方法对NiCo双原子团簇催化甲烷干法重整反应的体系进行了计算研究。通过计算结果得出甲烷脱氢、二氧化碳活化、C*和CH*的氧化、H2 和H2 O的生成四个反应过程可能的反应路径。最后,运用能量跨度模型分析循环反应的动力学信息,发现298K时甲烷脱氢过程中不易生成C*。913 K时甲烷脱氢过程决速中间体由IM1-1变成了IM6-1、决速过渡态由TS78-1变成了TS56-1;虽然可以生成C*,但能量跨度的减小加快了C*和CH*的消去。本工作可以了解NiCo双原子团簇催化甲烷干法重整的作用机理,为实验研究提供理论基础。
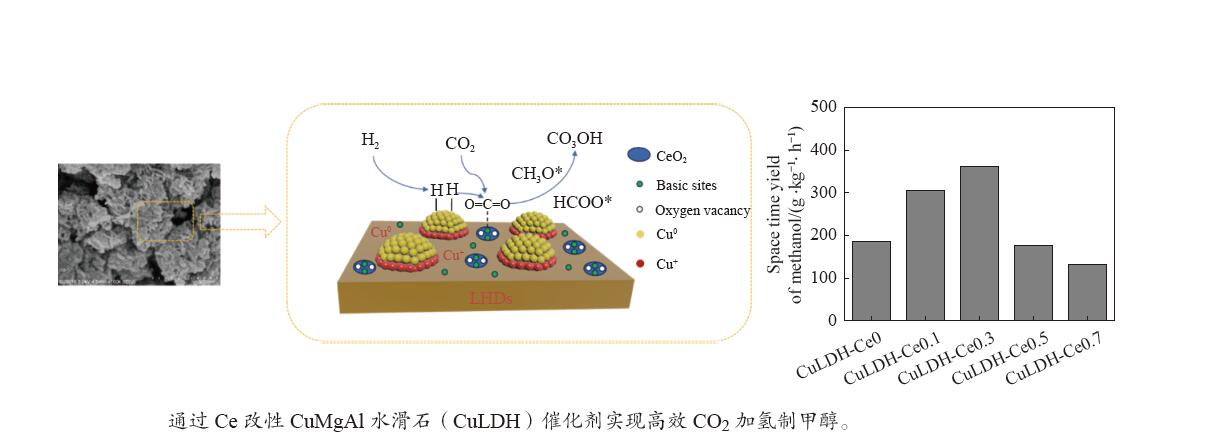
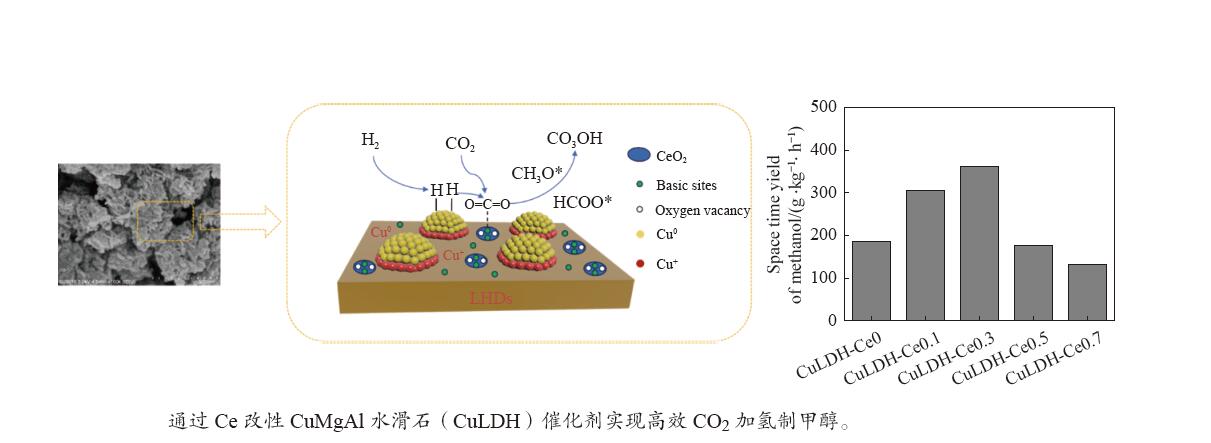
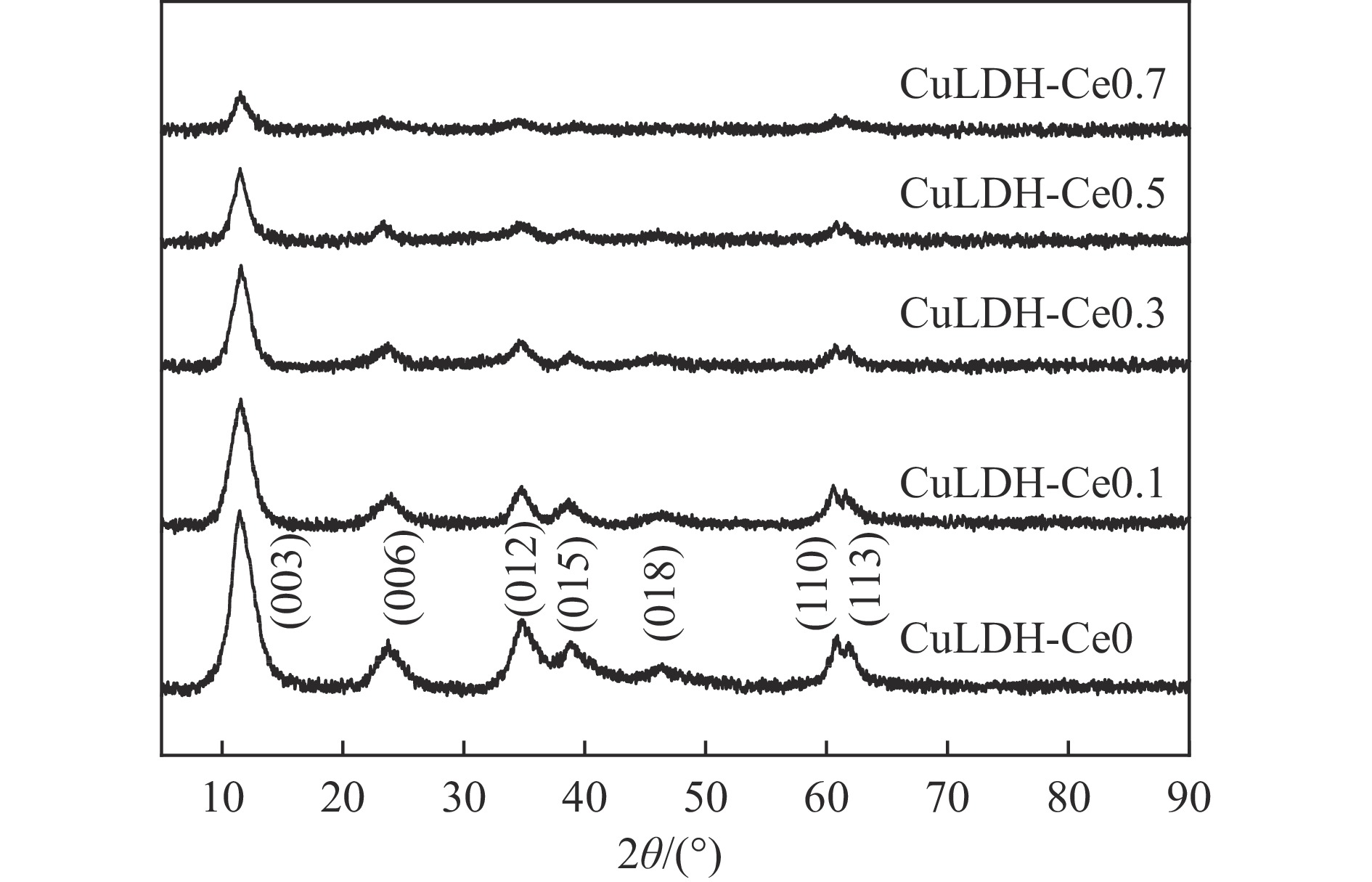
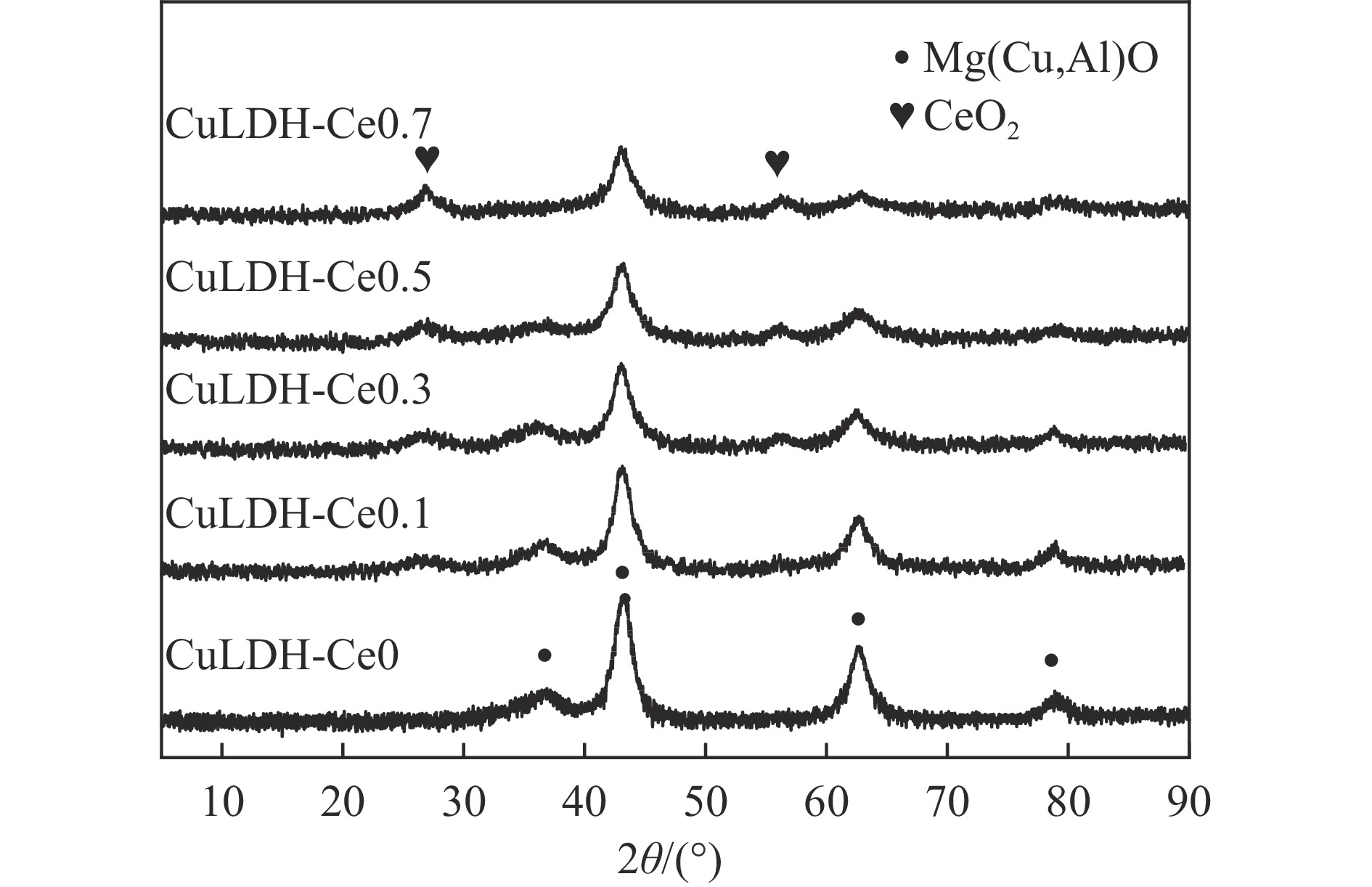
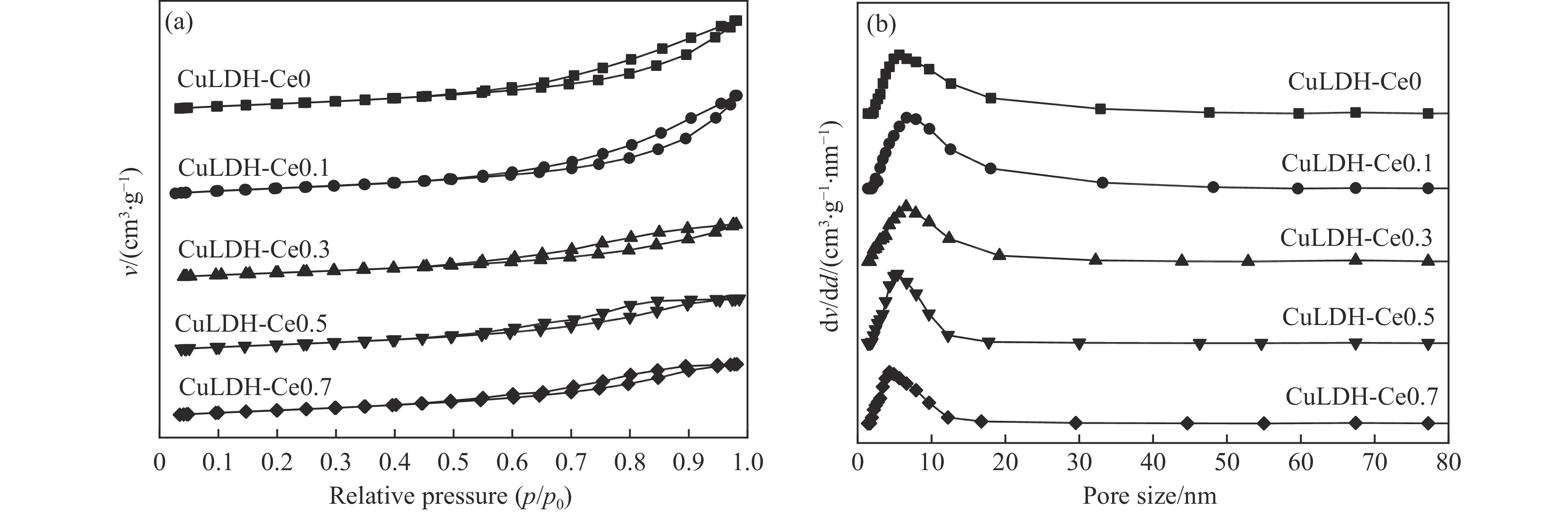
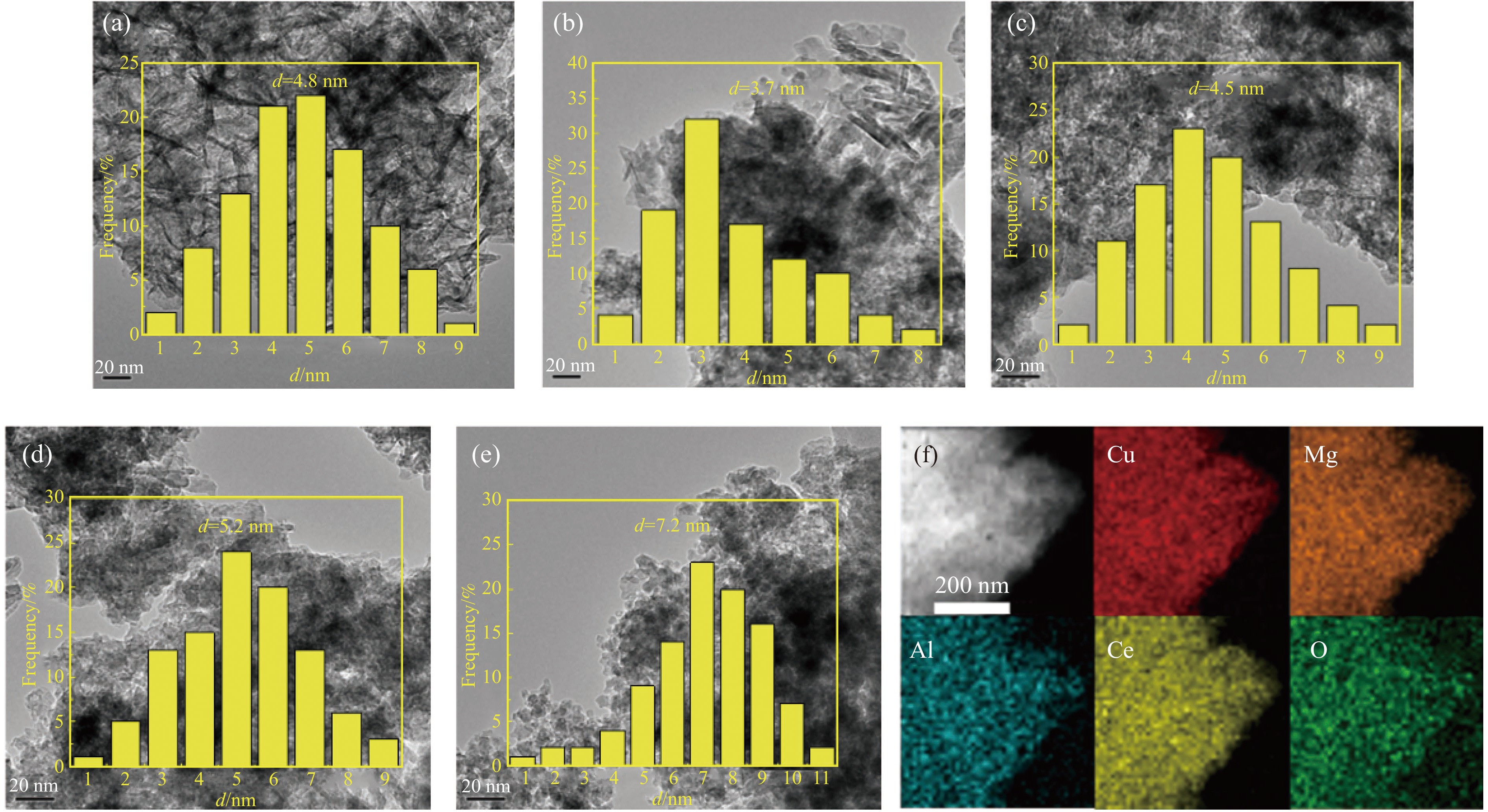
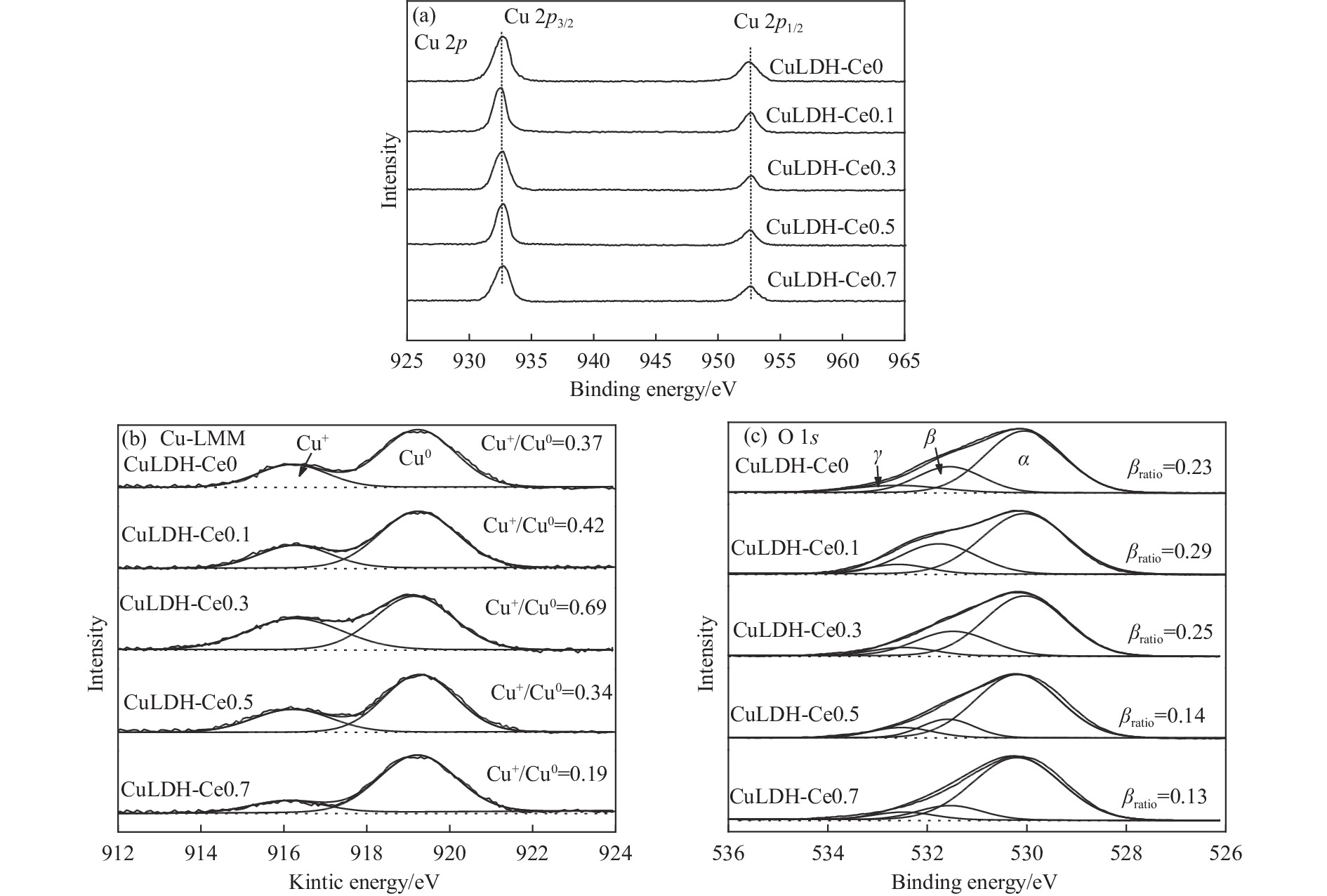
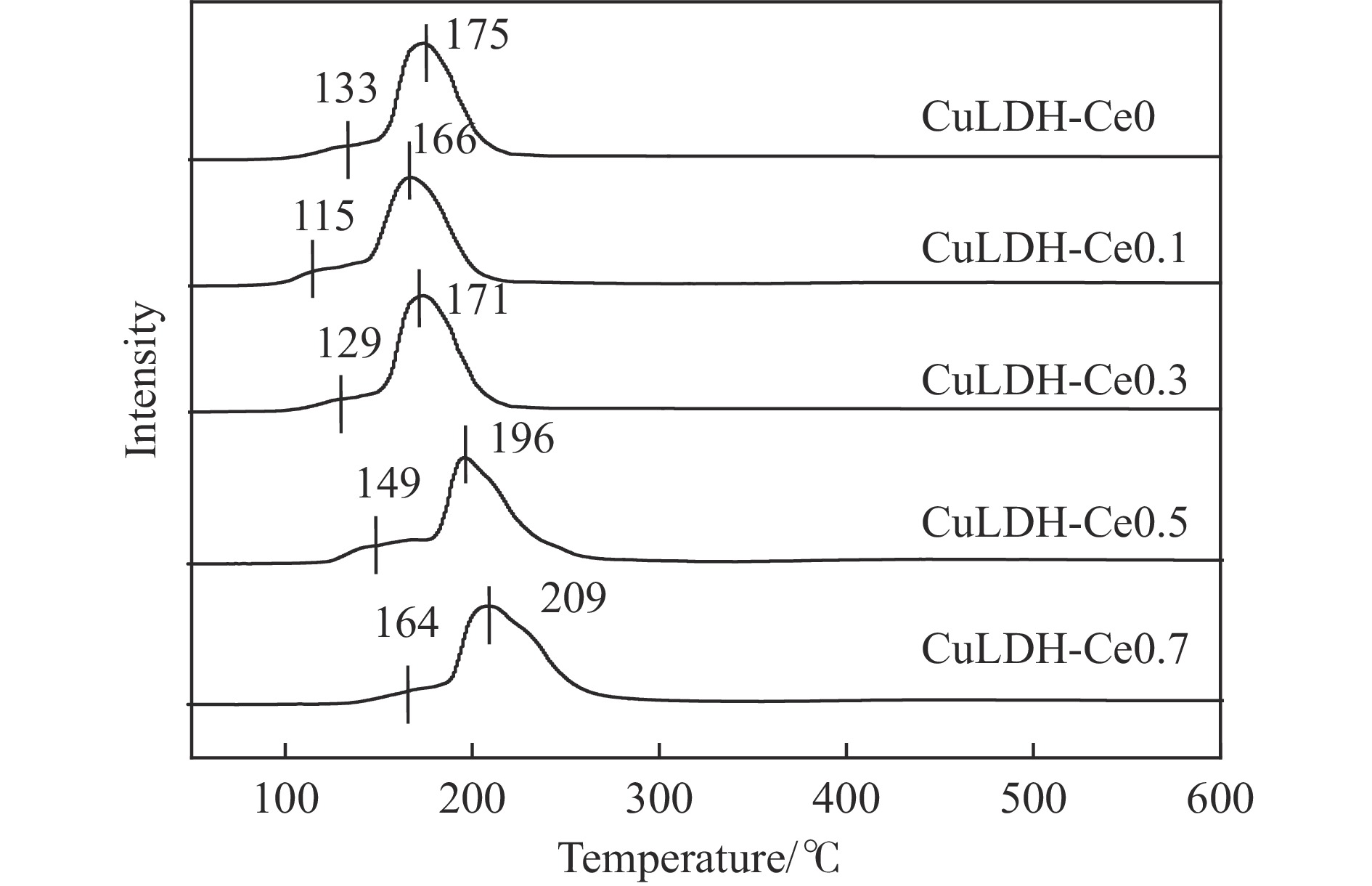
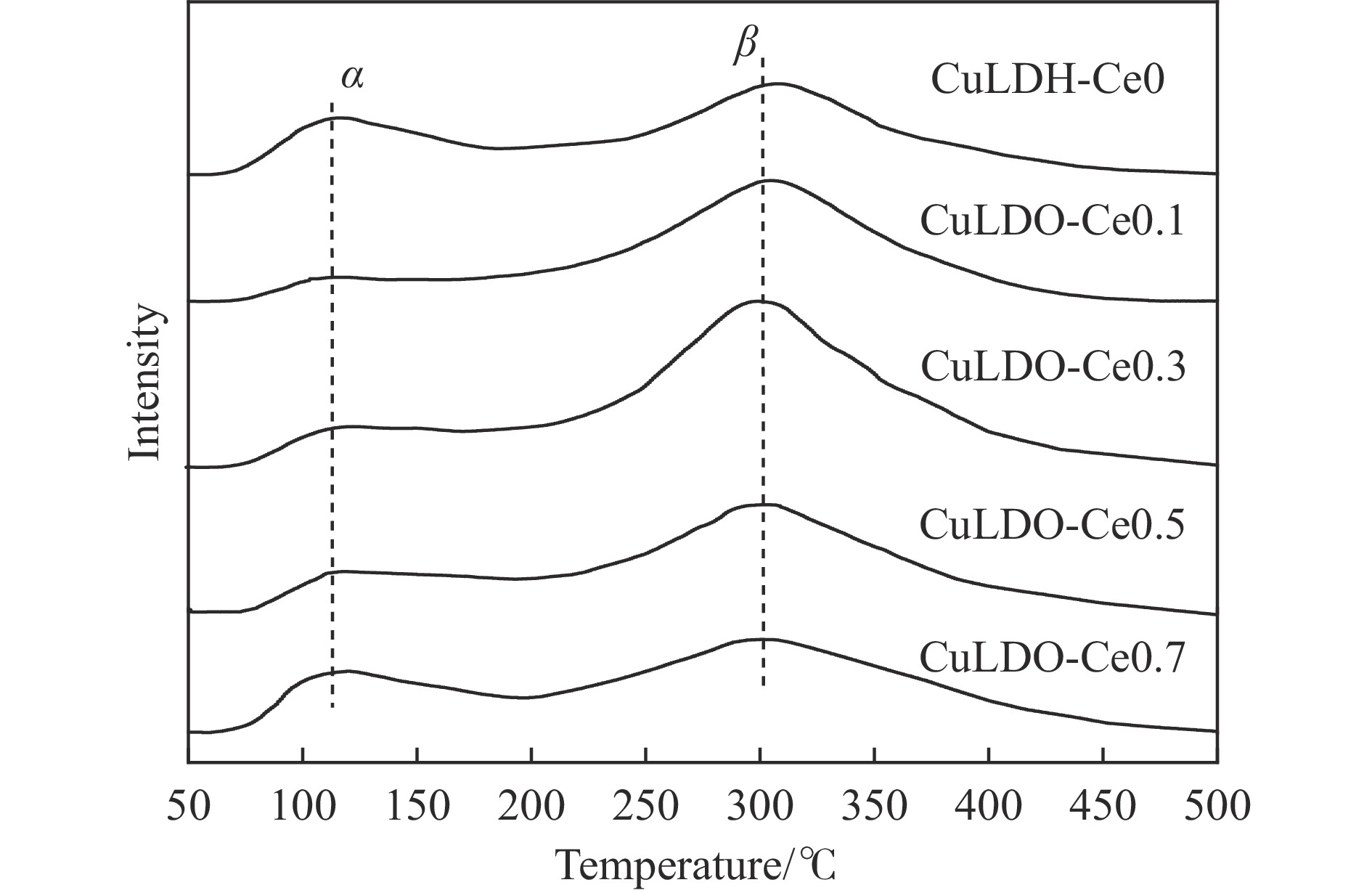
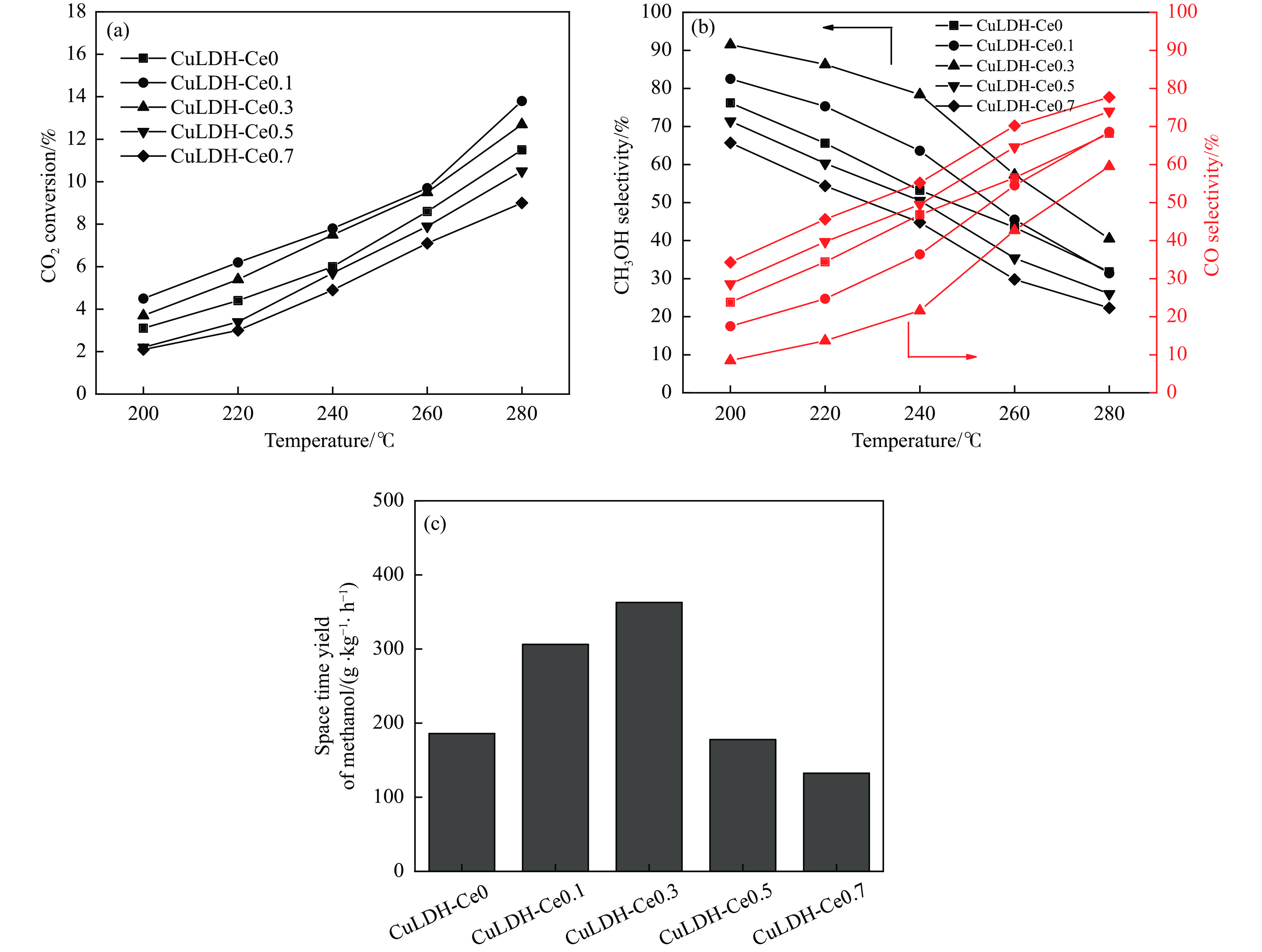
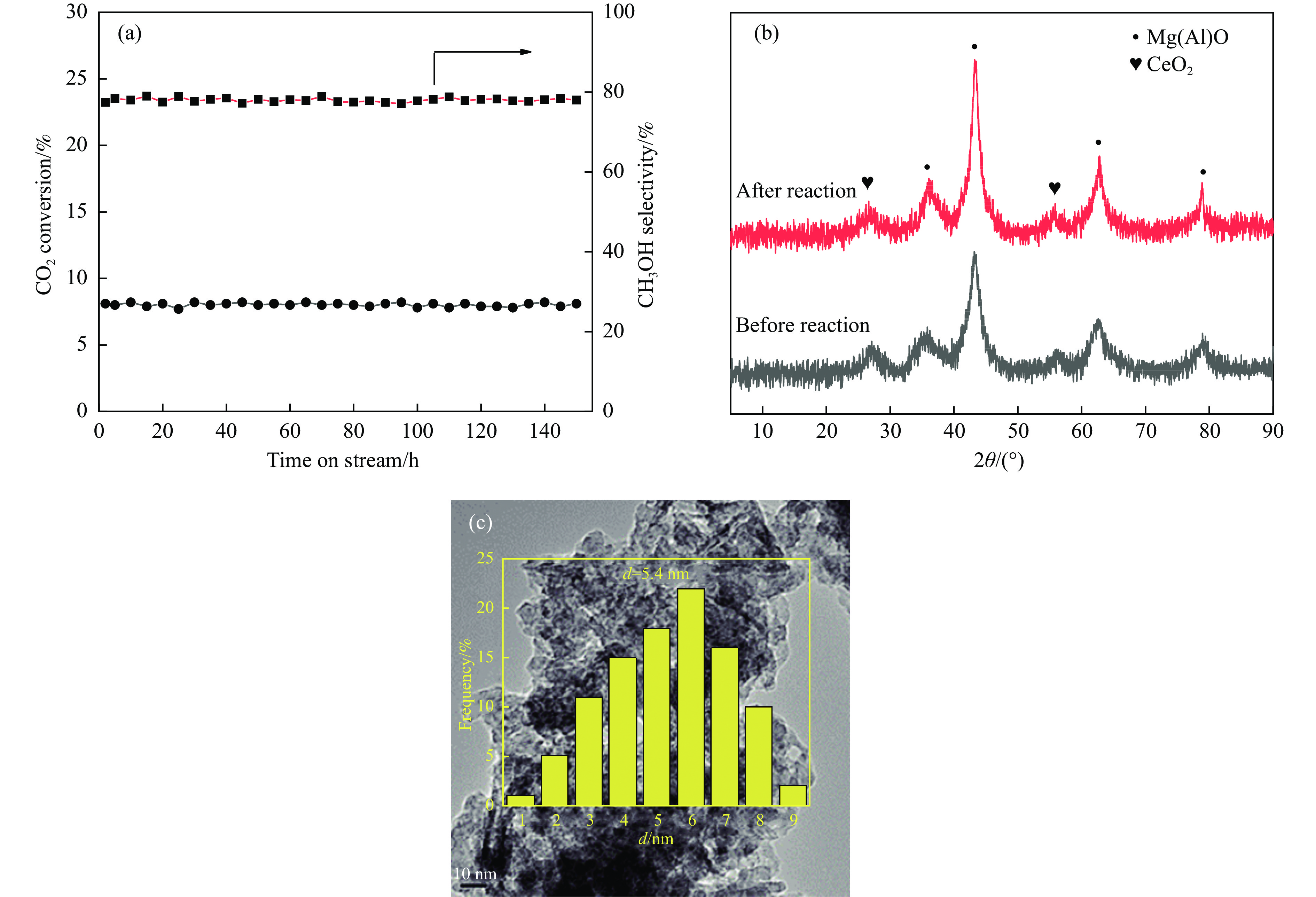
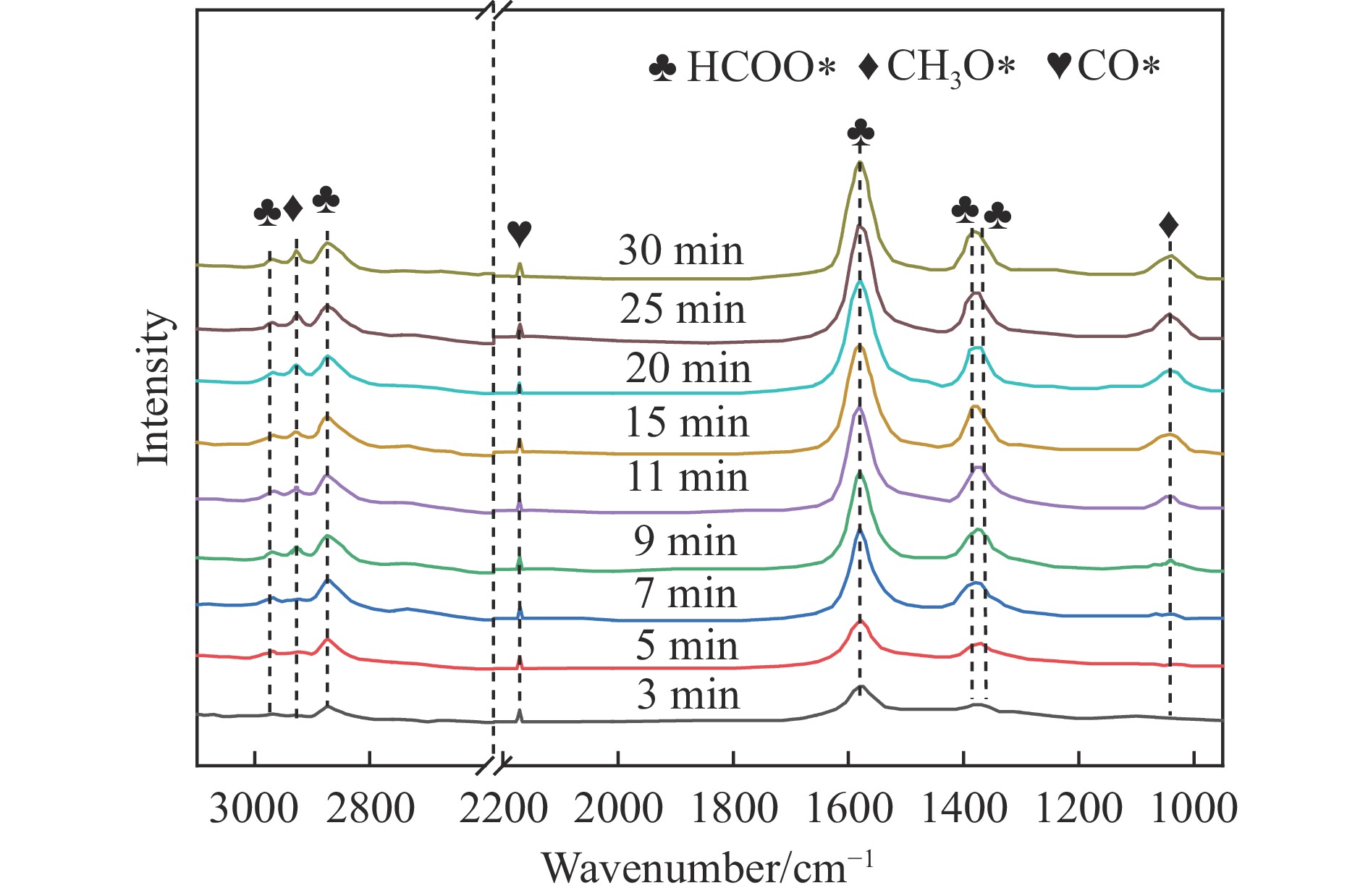
摘要: 通过向CuMgAl水滑石(CuLDH)催化剂中添加不同量的Ce,合成了一系列Ce改性的CuLDH-Cex 2 吸附-脱附(BET)、透射电子显微镜(TEM)、X射线光电子能谱(XPS)等分析手段对催化剂的理化性质进行表征。结果表明,添加Ce会改变Cu-LDH催化剂的水滑石结构,适量的Ce会增大催化剂的比表面积,改善了Cu颗粒的分散度。同时,适量的Ce有利于增加催化剂表面强碱性位点的密度和氧空位的数量,促进了CO2 的吸附和转化。Ce有利于调变催化剂表面的Cu+ /Cu0 比例,较高的Cu+ /Cu0 比例有利于甲醇的生成。当Ce/Cu比例为0.3时,在空速为9000 mL/(g·h),温度为240 ℃,压力为2.5 MPa的条件下,催化剂的CO2 的转化率为7.5%,甲醇选择性为78.4%,甲醇的时空收率最高可达362.8 g/(kg·h)。通过原位红外光谱(in-situ DRIFTS)证明CuLDH-Ce0.3催化剂在CO2 加氢合成甲醇过程中遵循HCOO*反应路径。
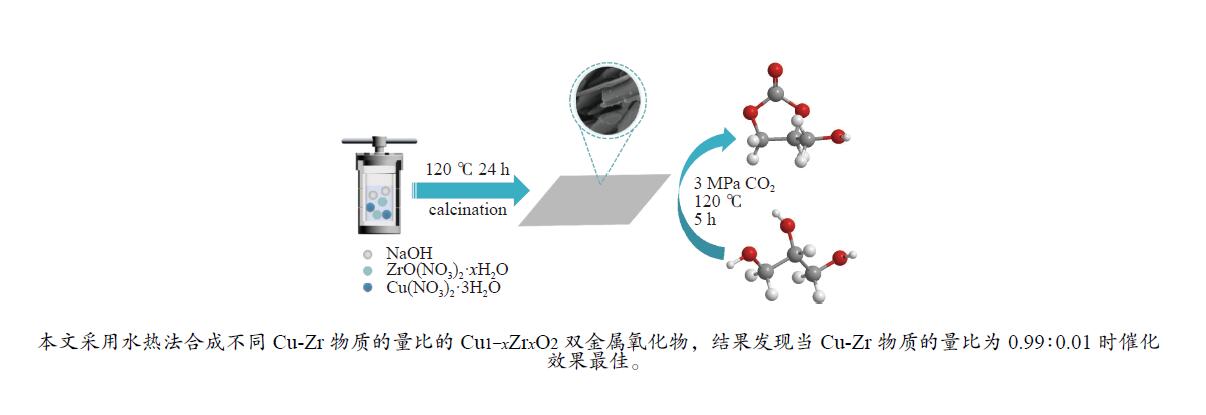
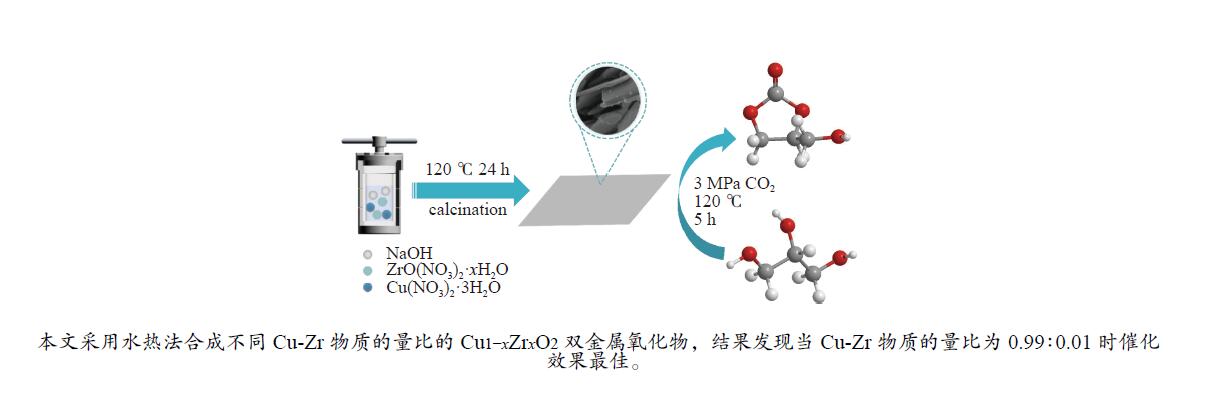
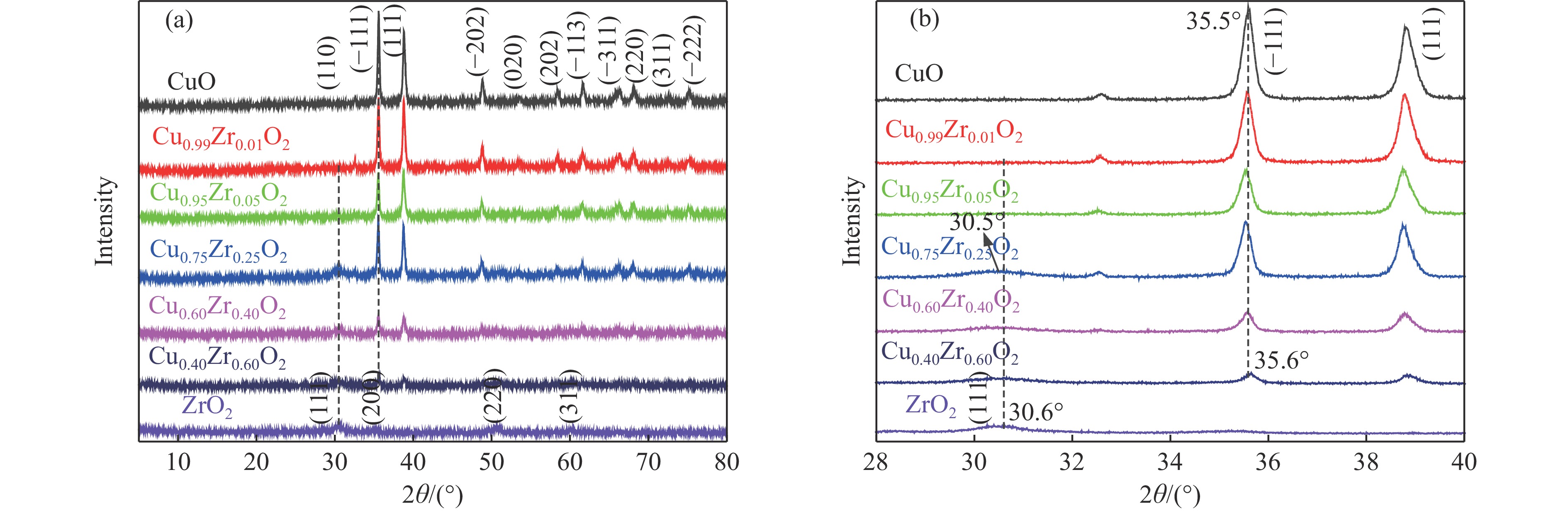
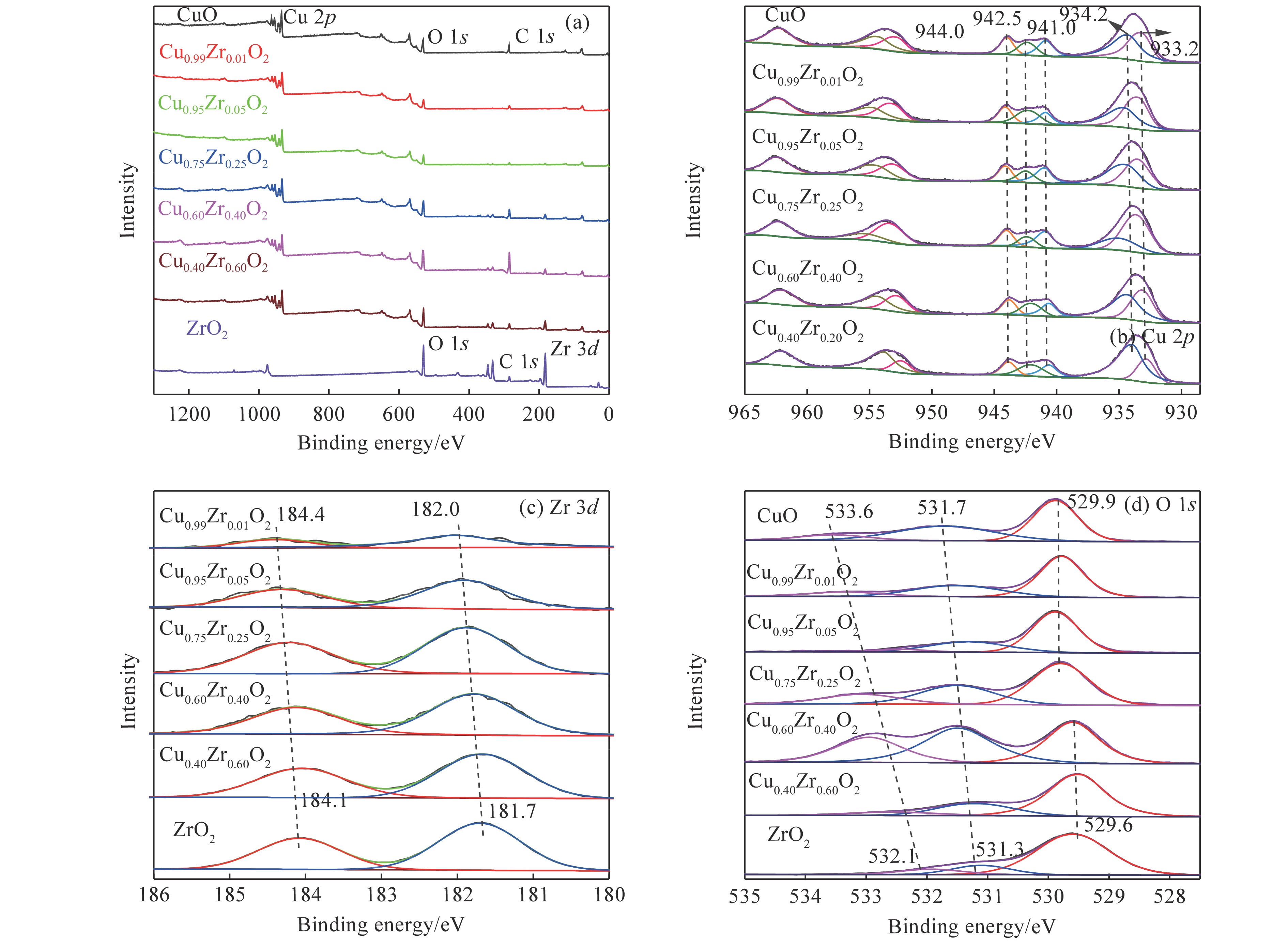
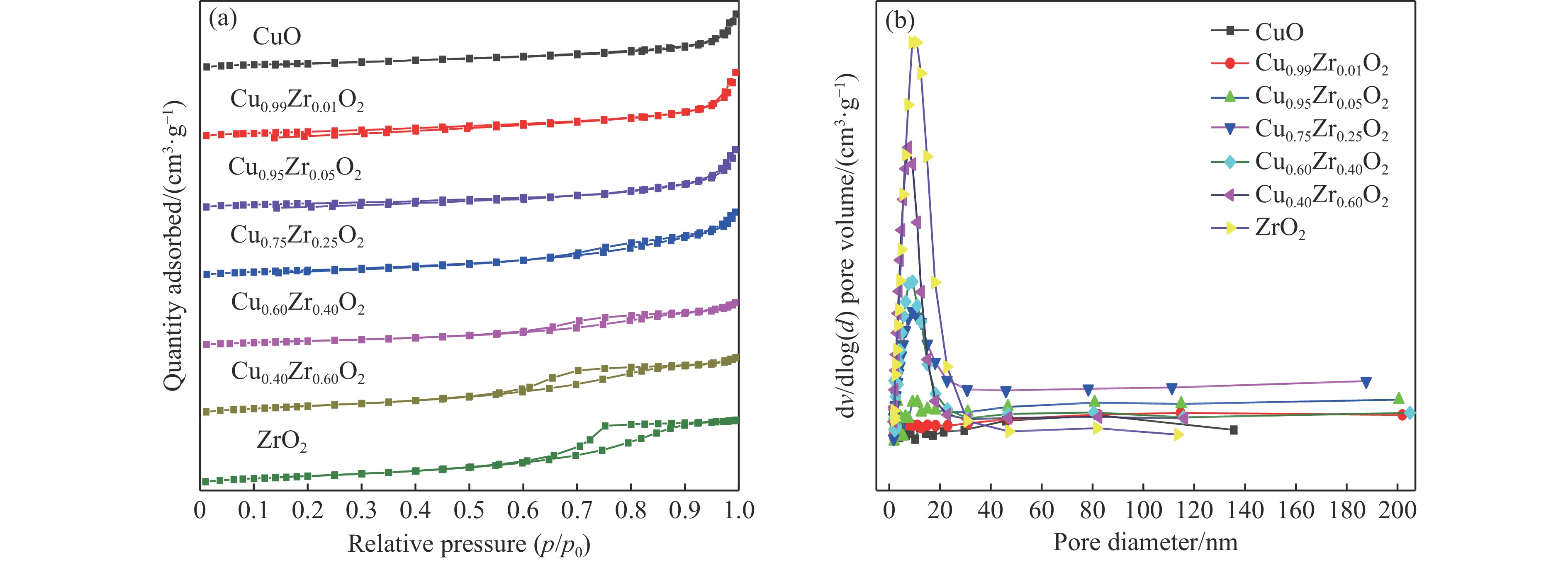
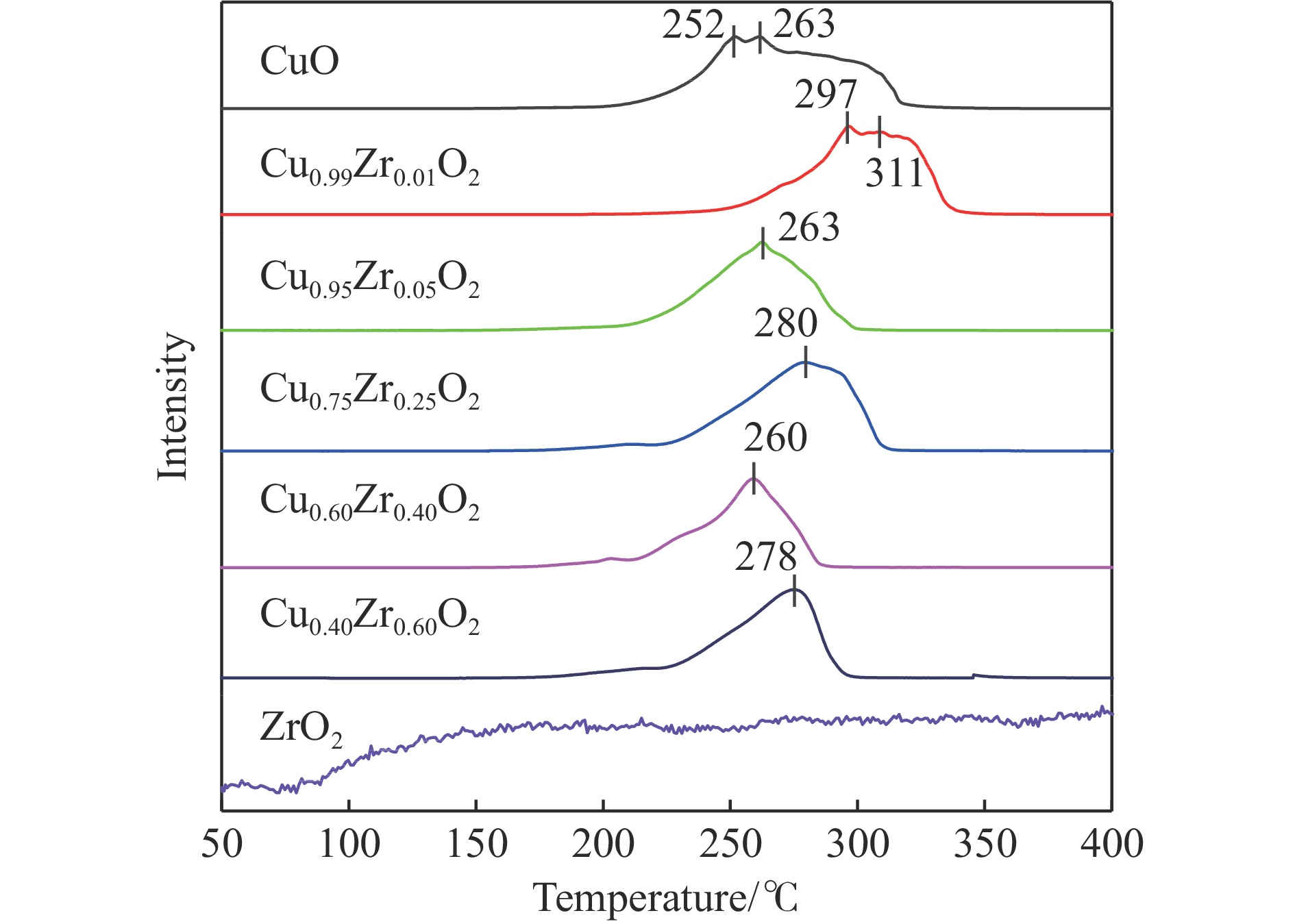
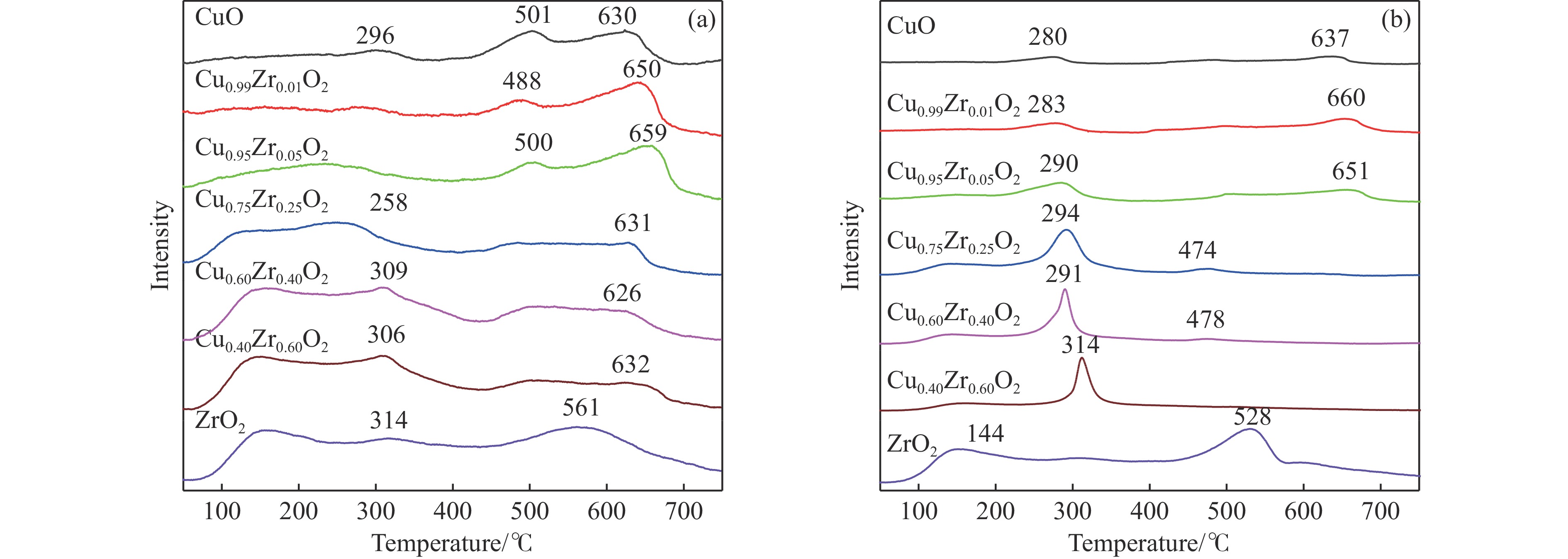
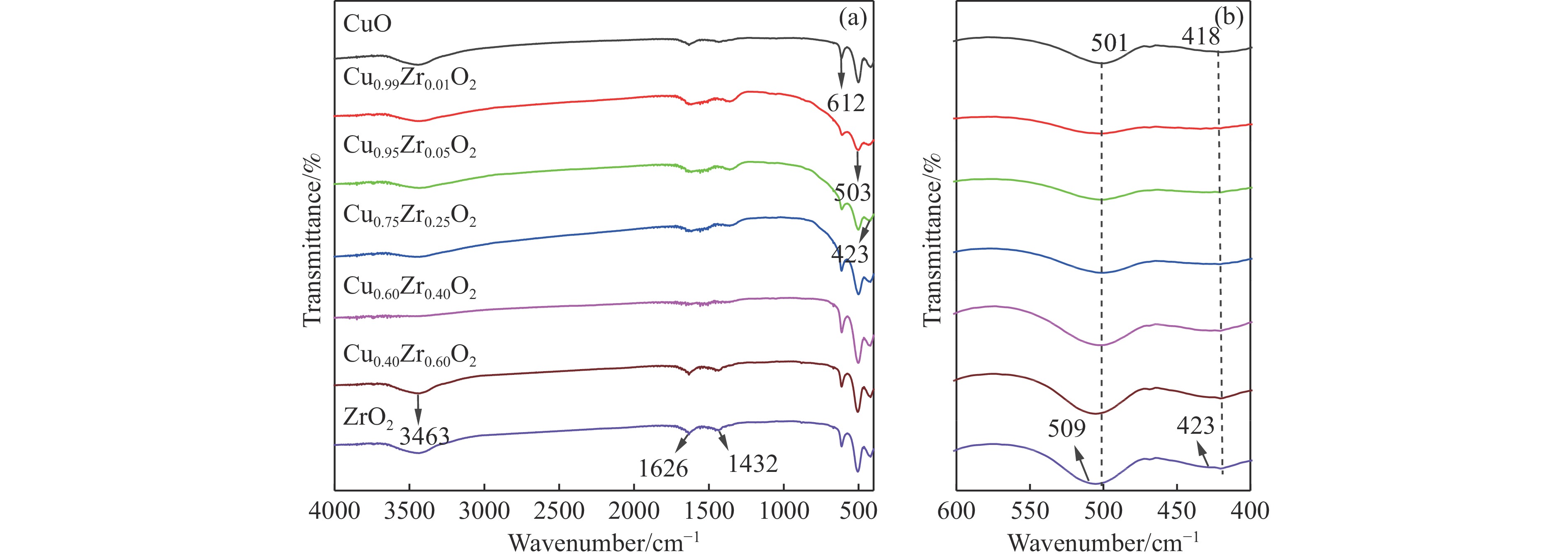
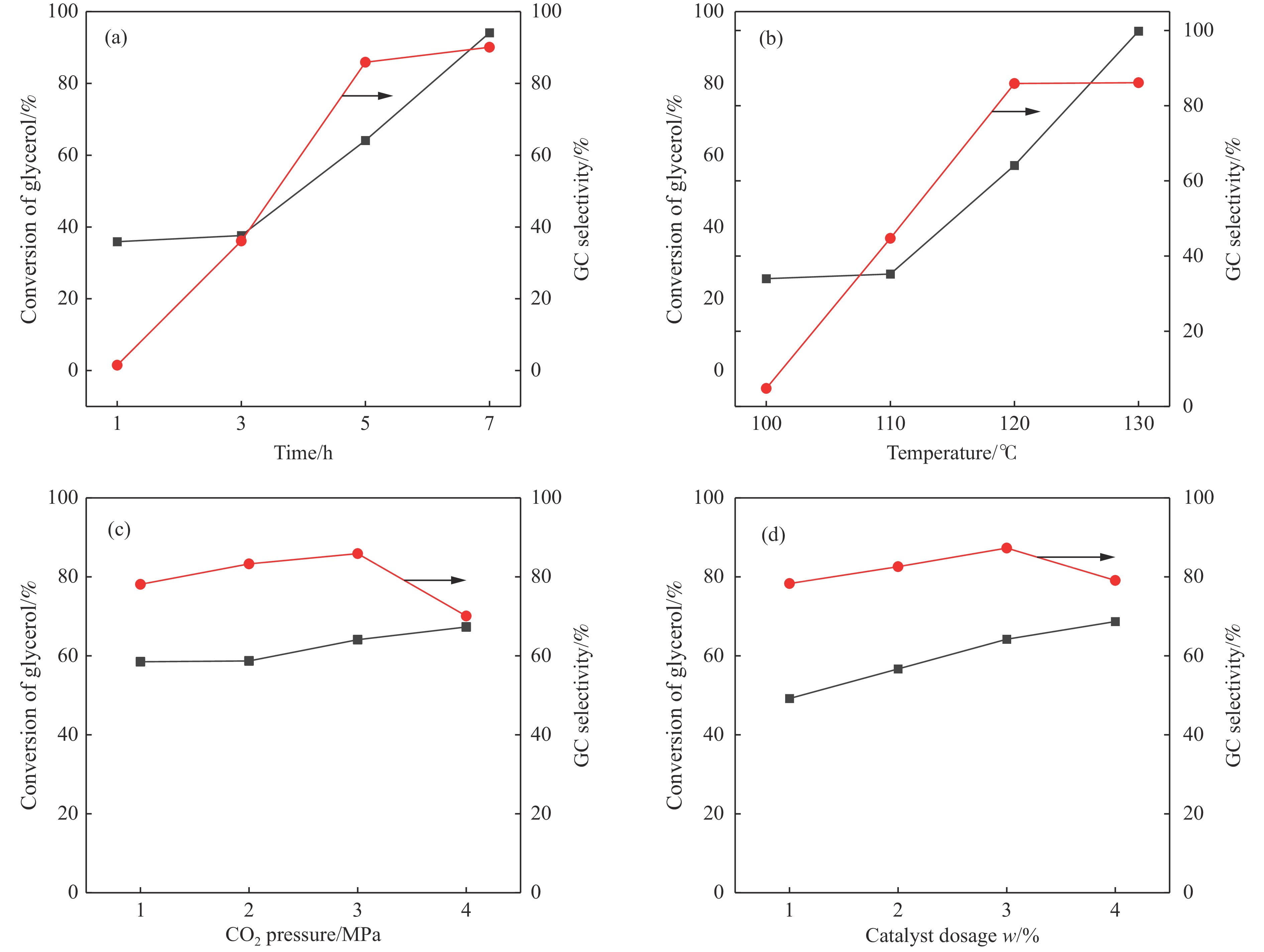
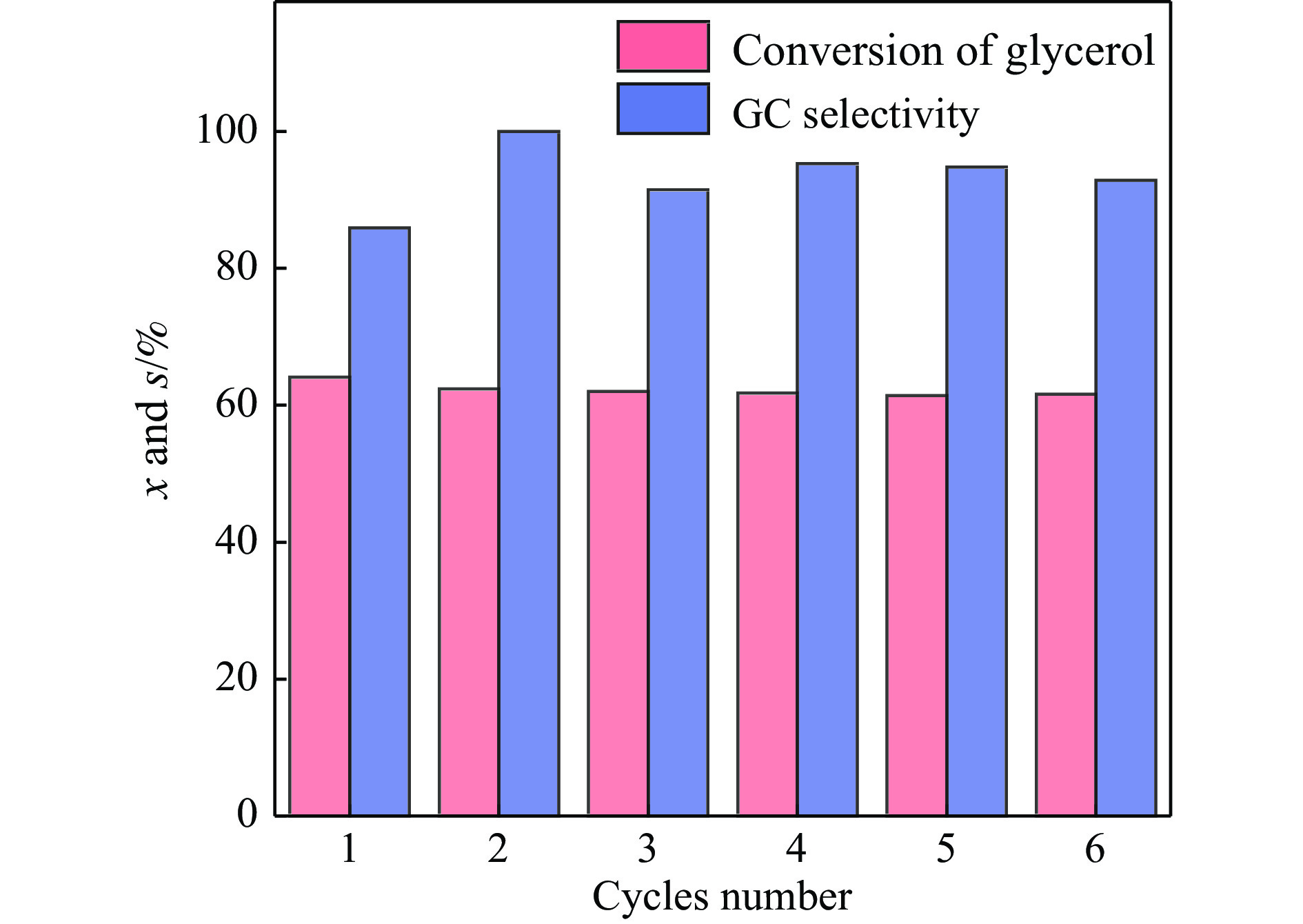
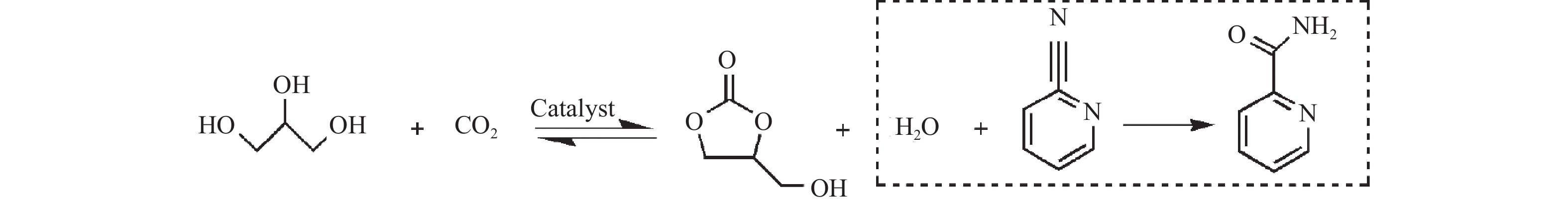
摘要: 采用水热法合成了一系列不同Cu-Zr物质的量比的Cu1−x Zrx 2 双金属氧化物,以此为催化剂,将生物柴油生产过程副产物甘油与温室气体CO2 耦合反应制备精细化工产品碳酸甘油酯。结果表明,Zr掺杂量不同,催化剂对甘油羰基化反应效果呈现明显差距,最佳反应条件下,Cu0.99 Zr0.01 O2 催化剂具有最佳催化性能,甘油转化率和碳酸甘油酯选择性分别达到64.1%和85.9%。并且发现与纯CuO和纯ZrO2 相比,Cu1−x Zrx 2 复合氧化物在甘油与CO2 耦合反应体系中表现出更强的催化活性,结合X射线粉末衍射(XRD)、扫描电子显微镜(SEM)、透射电子显微镜(TEM)、X射线光电子能谱(XPS)、N2 吸附-脱附、程序升温还原(H2 -TPR)、程序升温脱附(TPD)、傅里叶变换红外光谱(FT-IR)等表征手段,推测高活性与Zr在CuO表面的分散程度、催化剂表面氧物种含量及酸碱性位点数量有关。此外,为了研究催化剂的稳定性,以Cu0.99 Zr0.01 O2 催化剂作为基准进行了循环性能测试,结果表明,经过六次循环后,甘油的转化率和碳酸甘油酯的选择性未发生明显变化,说明该催化剂稳定性良好。
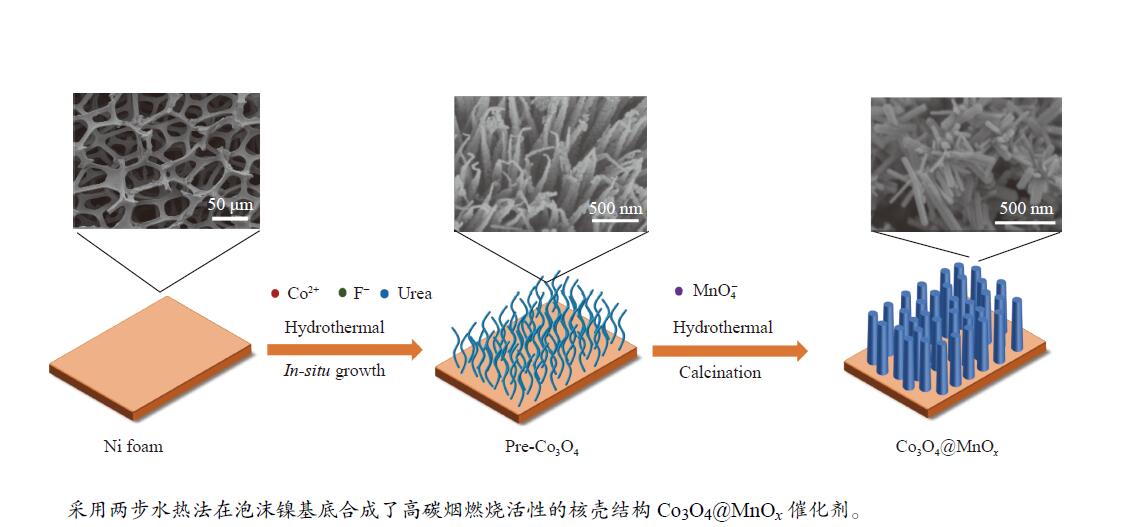
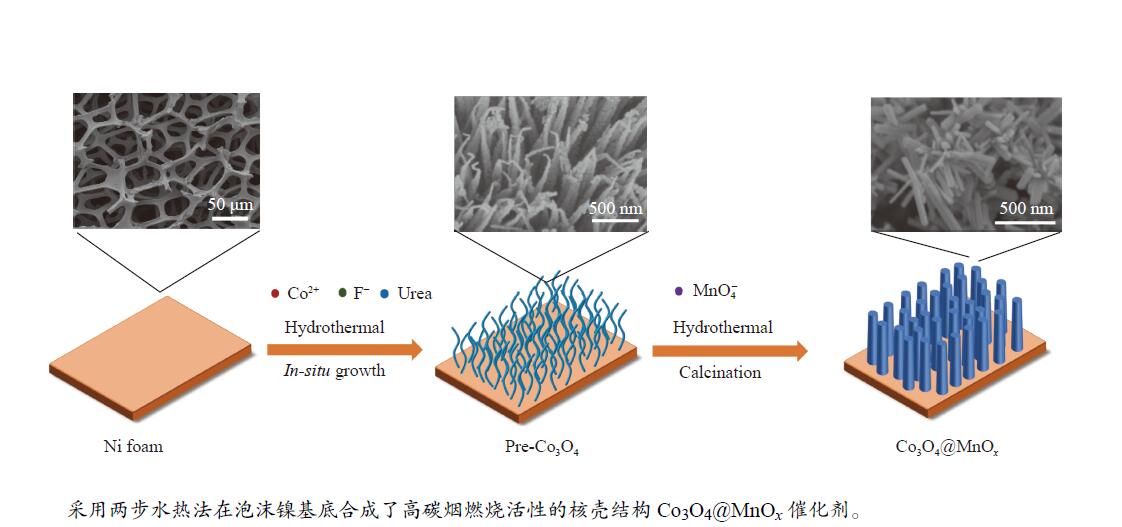
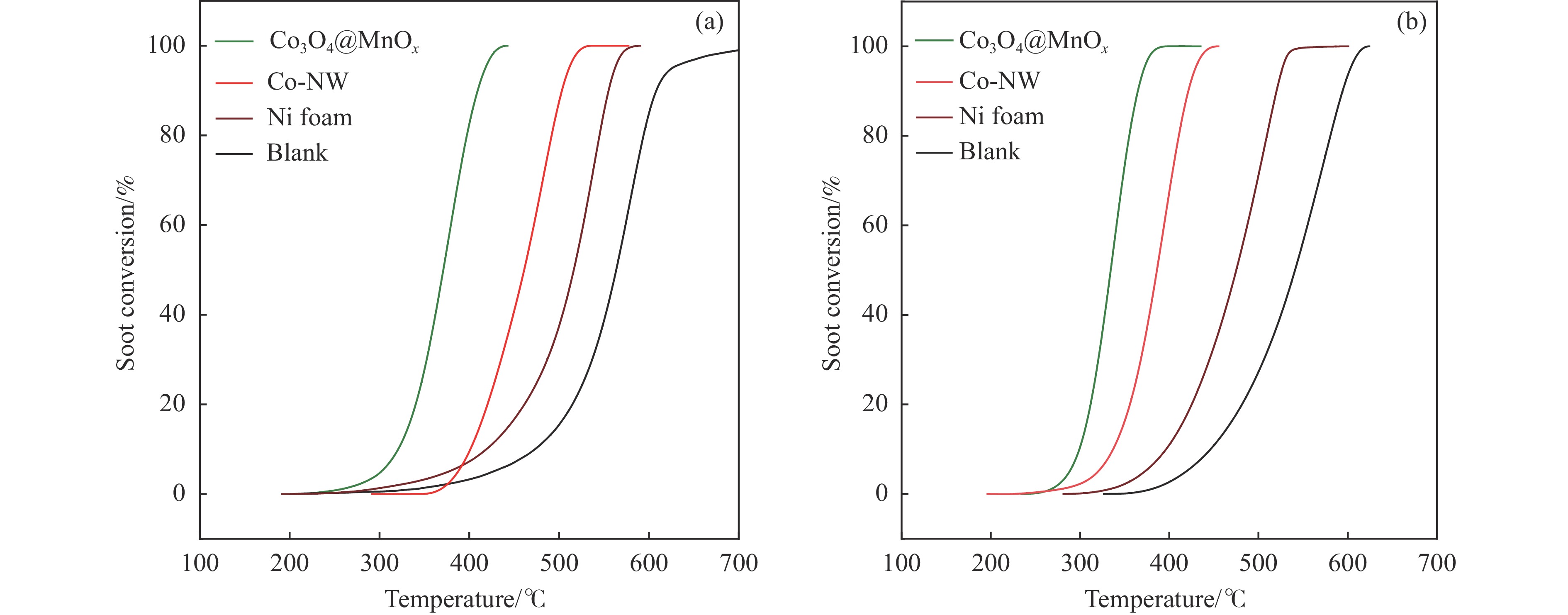
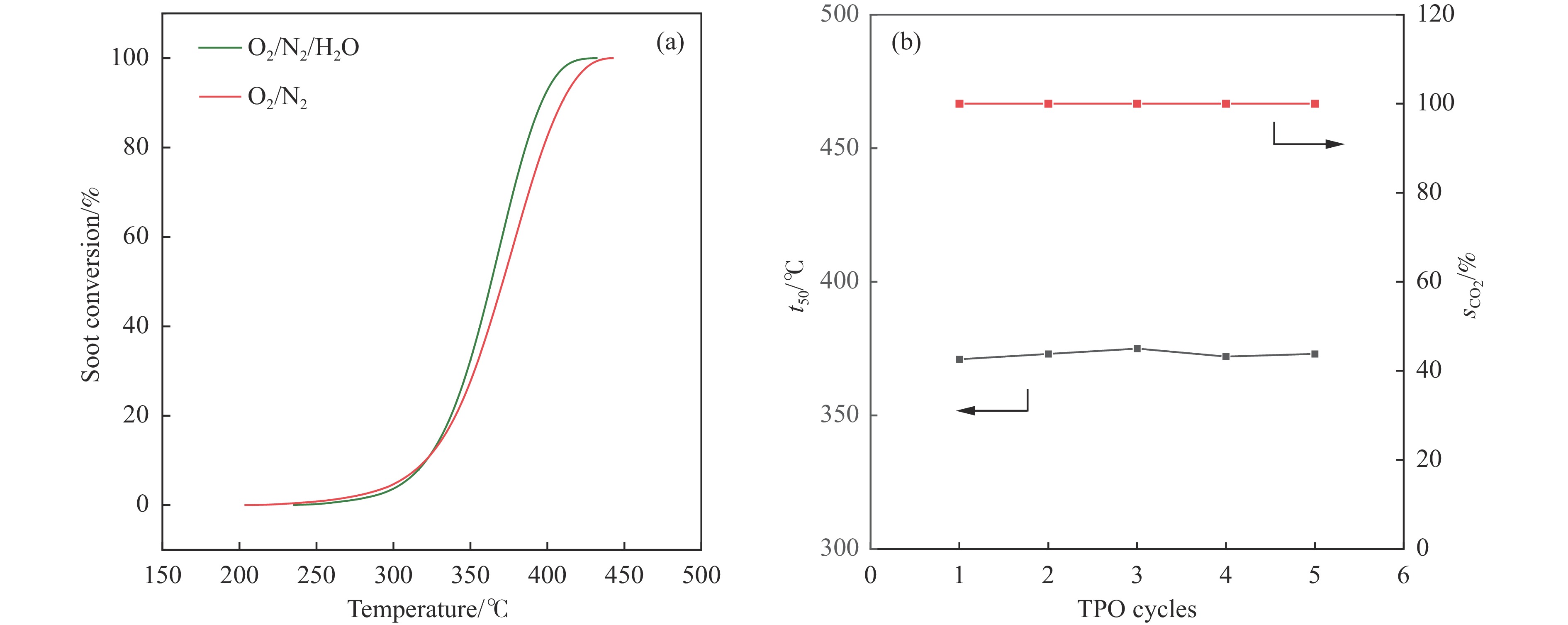
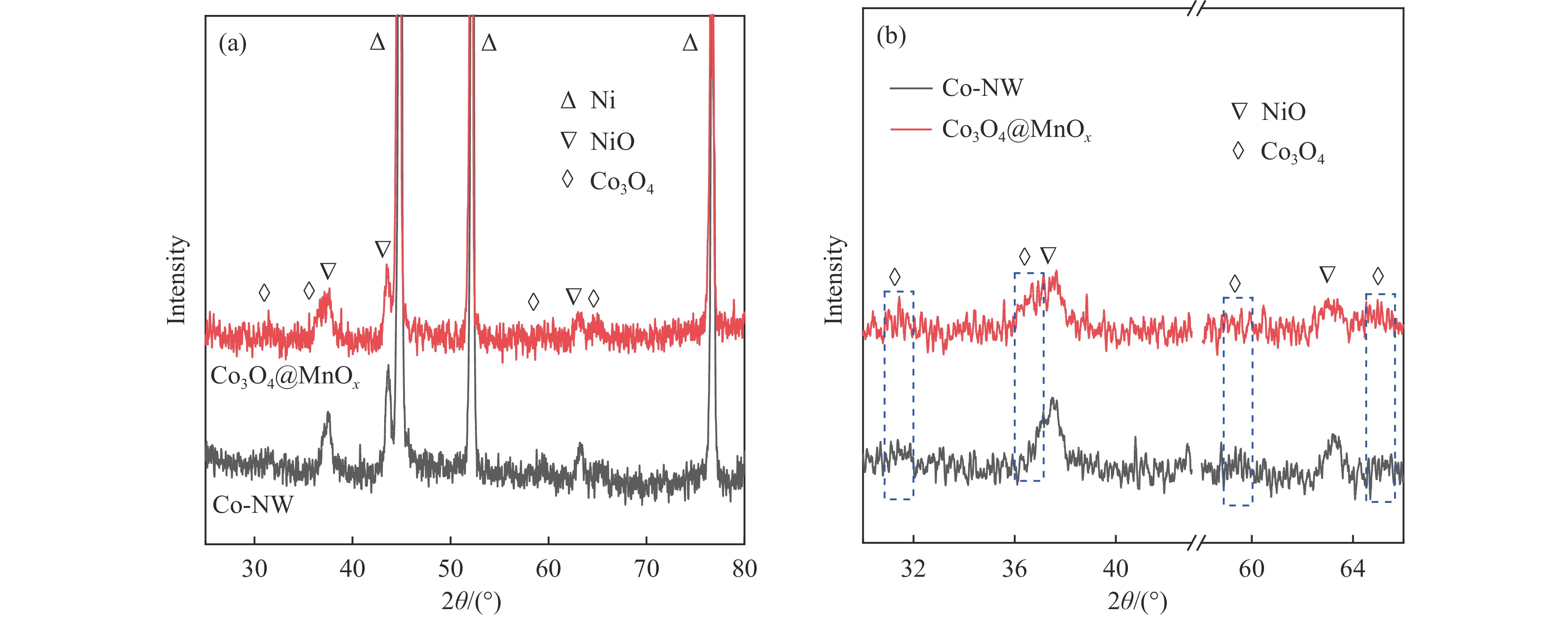
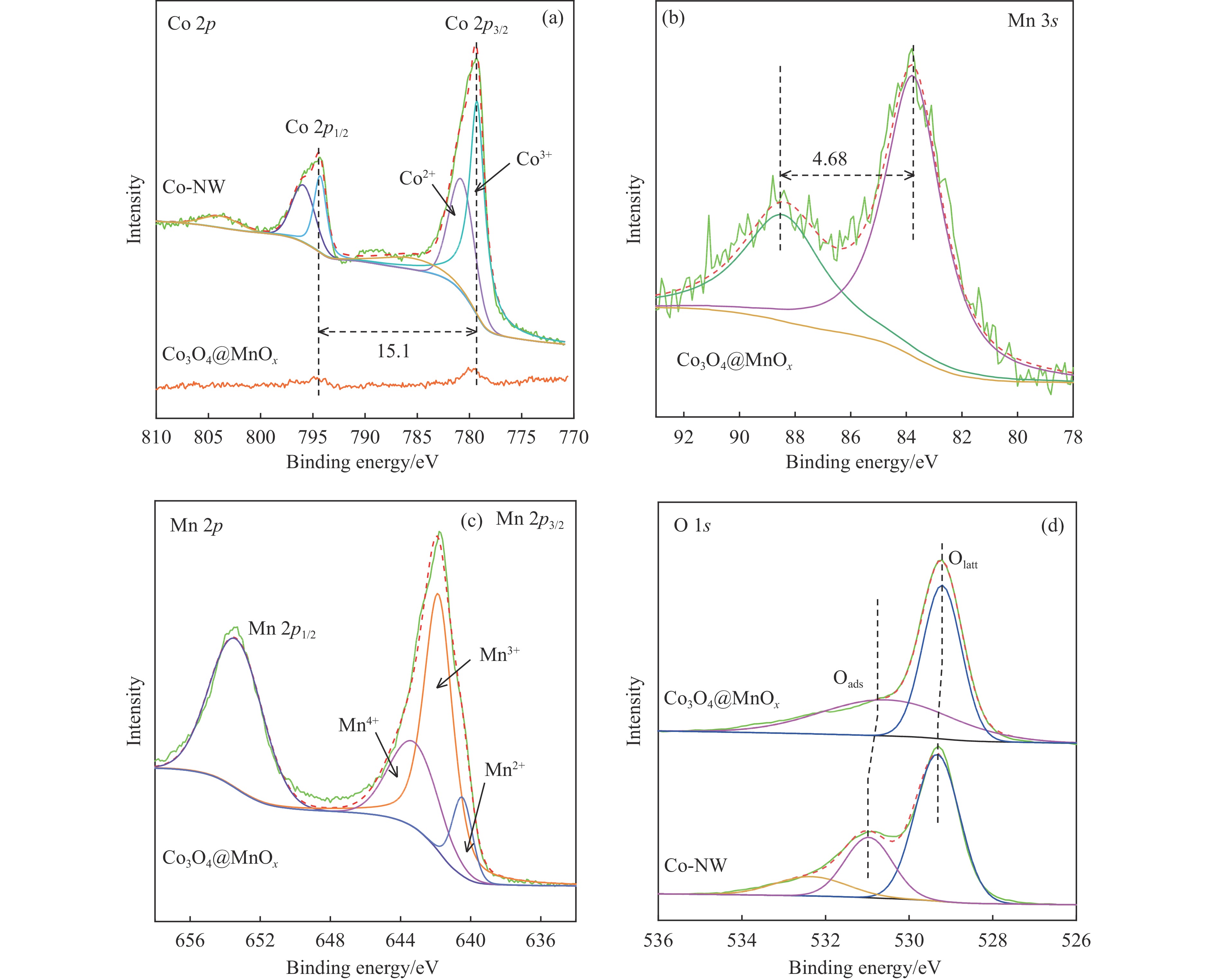
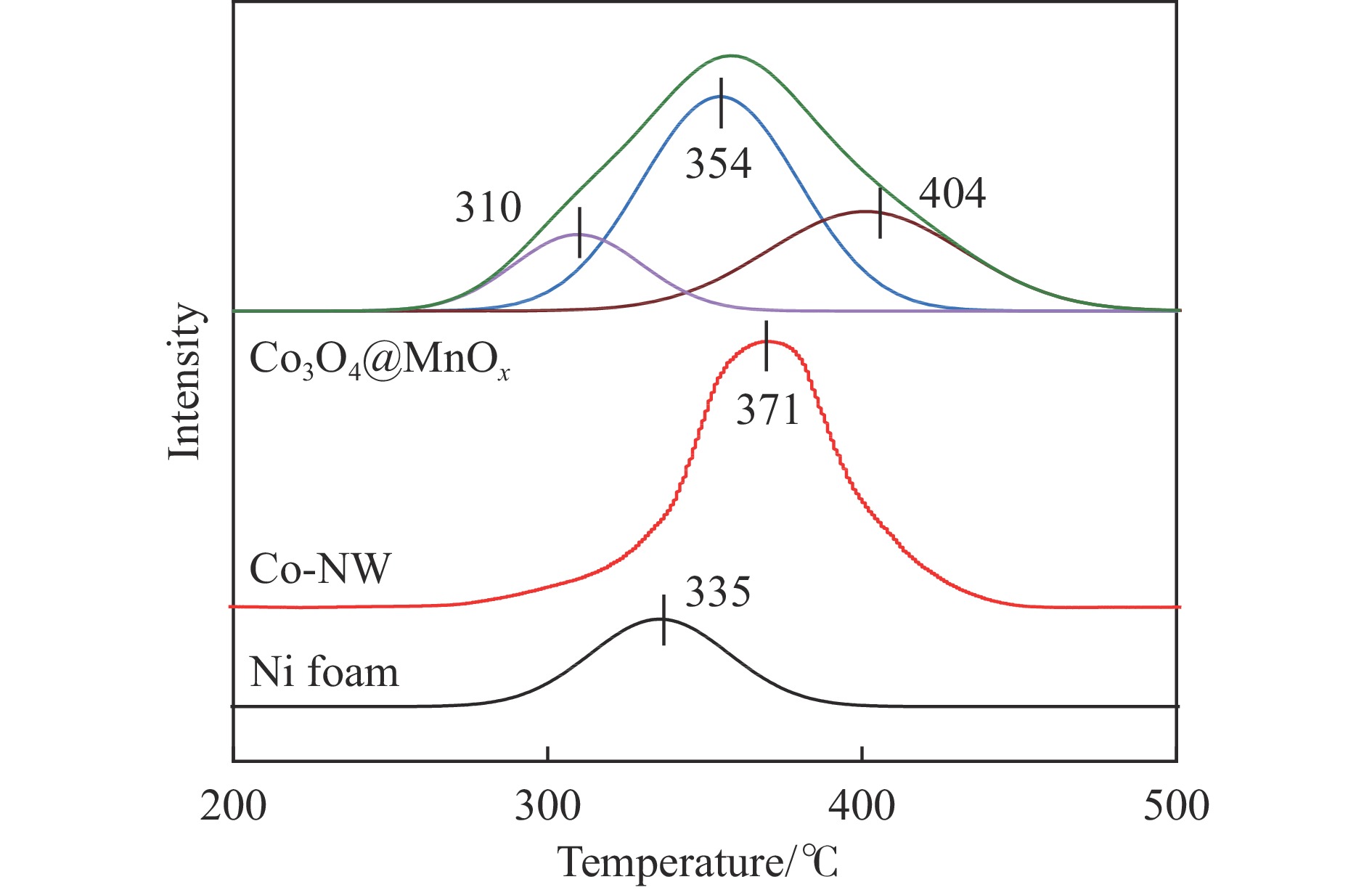
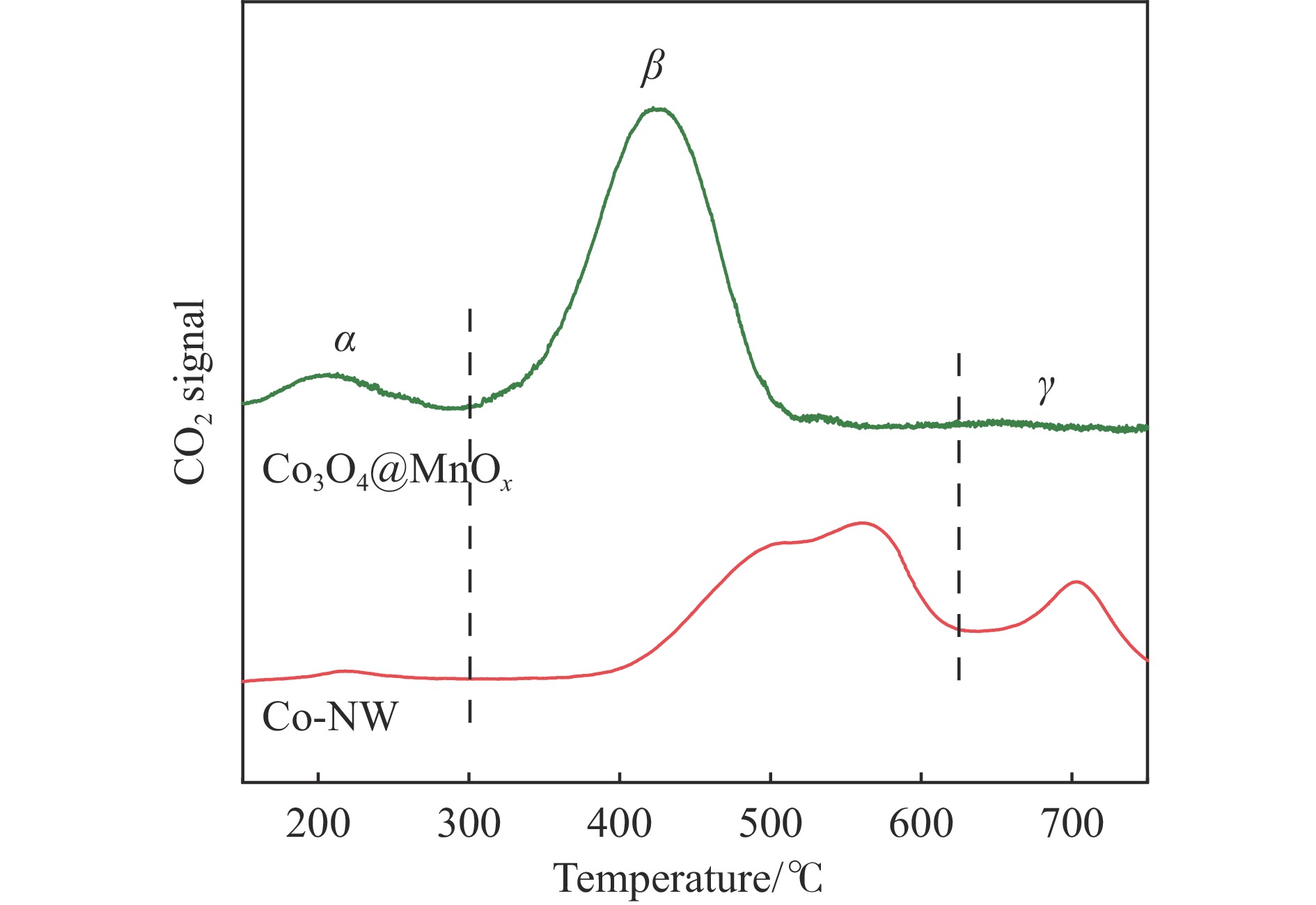
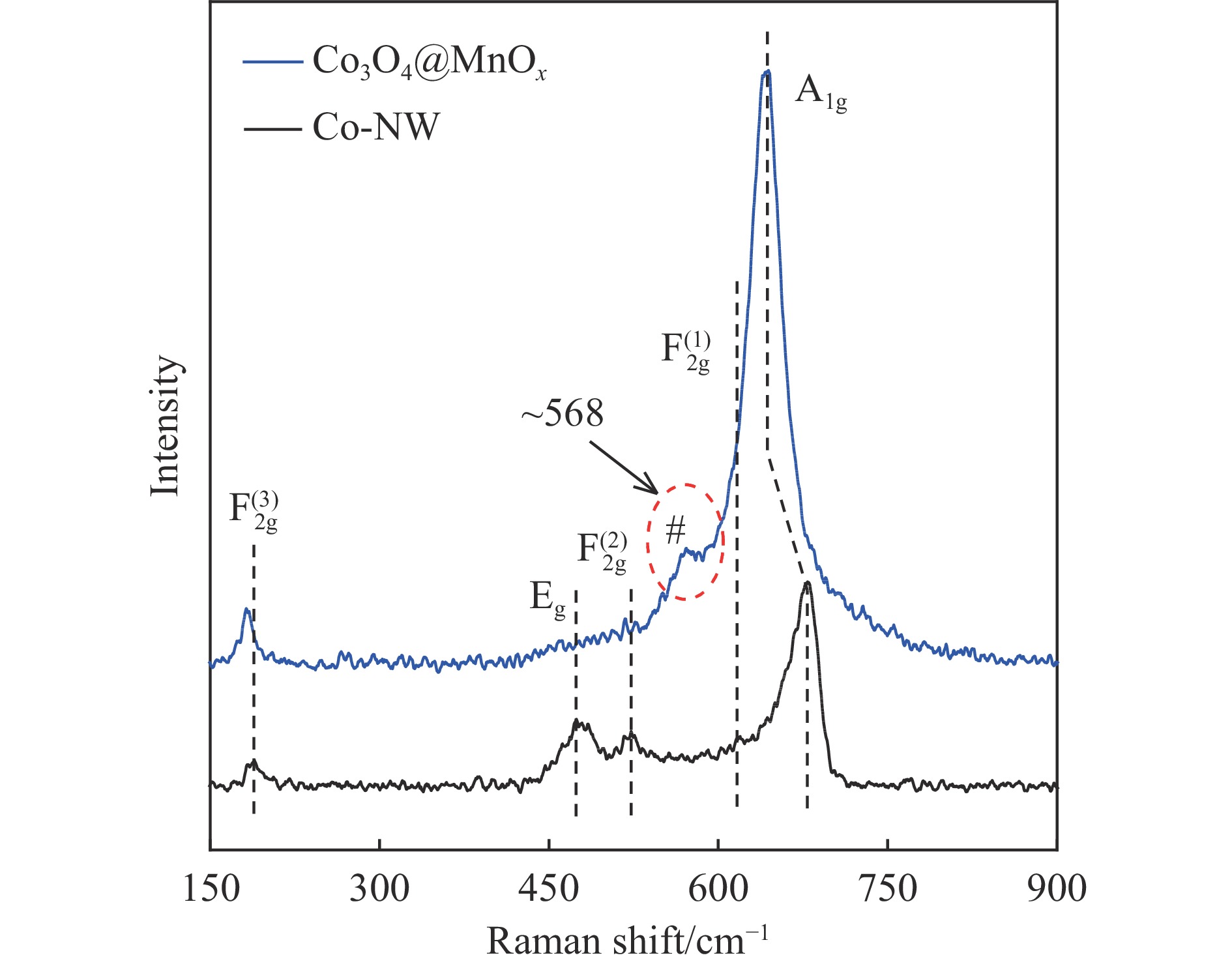
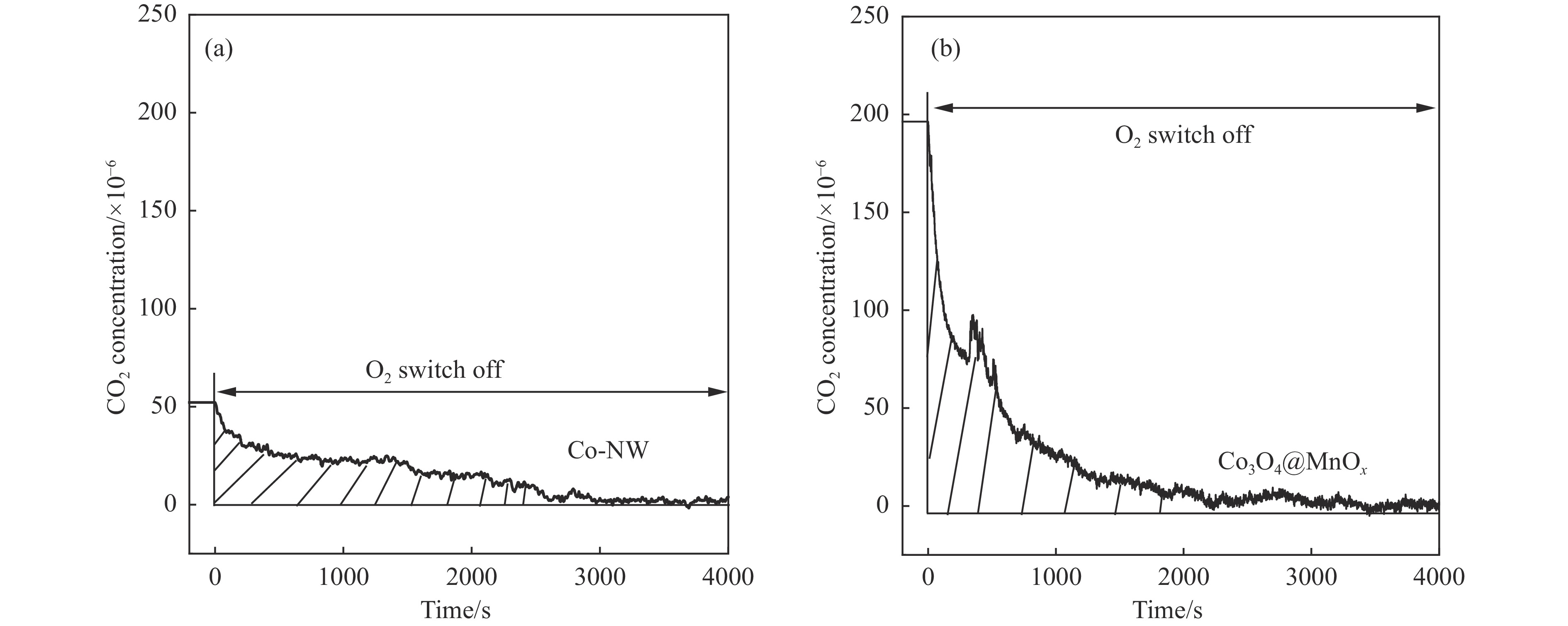
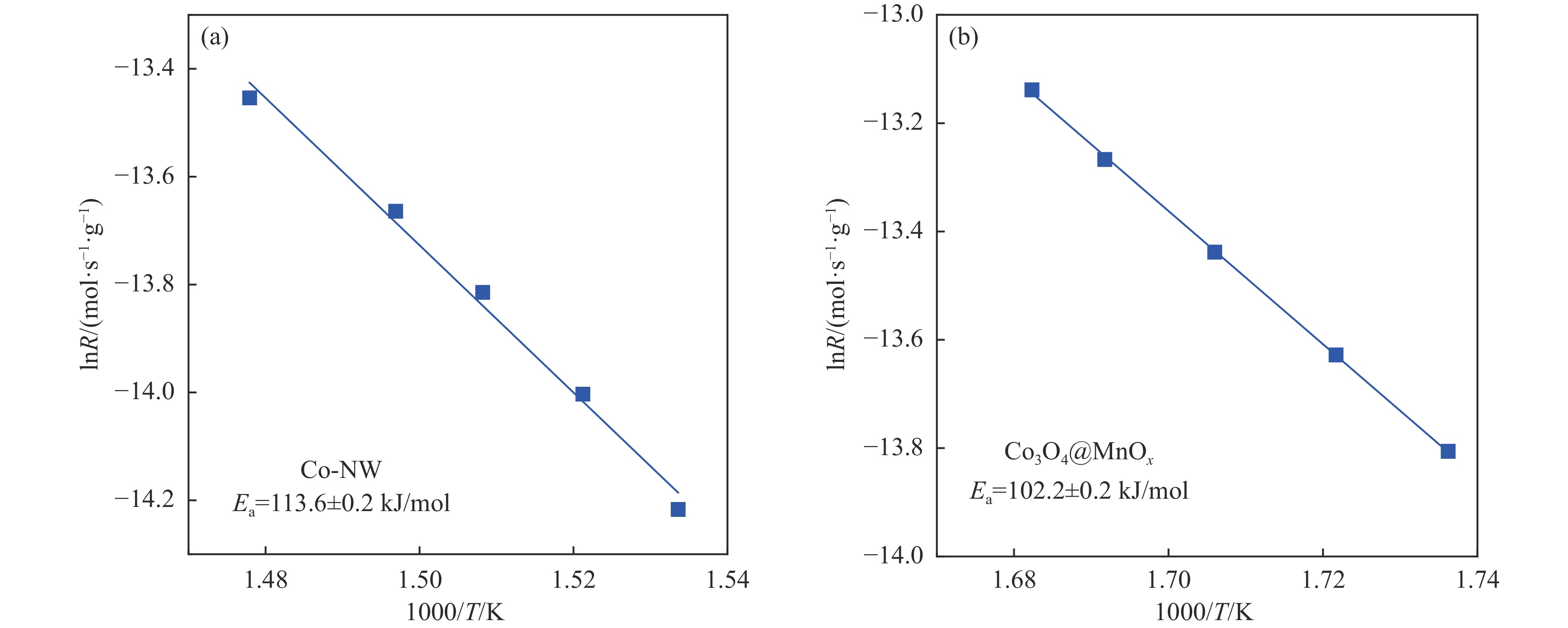
摘要: 采用两步水热法在泡沫镍基底上合成了具有纳米棒形貌的 Co3 O4 @MnOx 3 O4 @MnOx 3 O4 为核、以 MnOx 3 O4 @MnOx 3 O4 与 MnOx 4+ 和 Mn3+ 以及更多的表面氧空位,其氧化还原性能提高,催化剂的活性氧物种数量增加了两倍,催化性能得到改善,在 NO 存在的反应气氛中使碳烟起燃温度降低148 ℃。此外,相比催化剂 Co-NW,Co3 O4 @MnOx
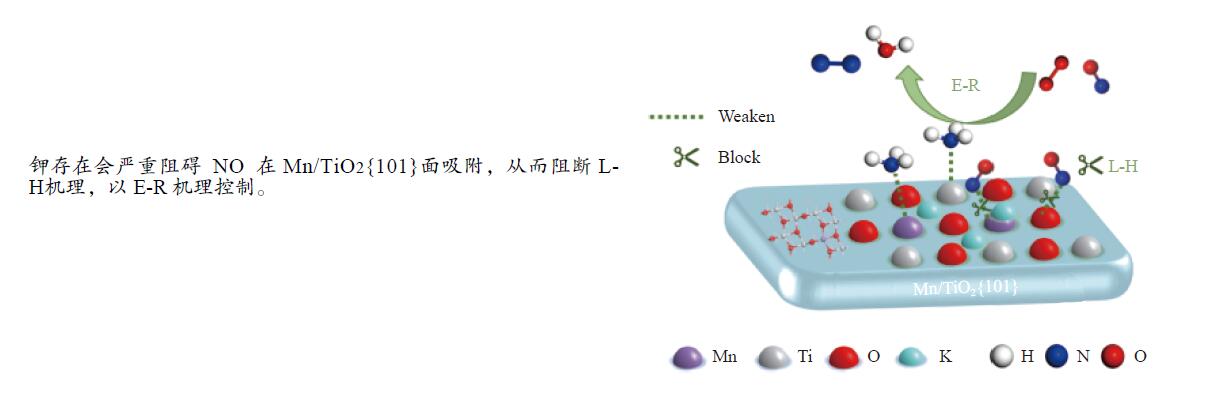
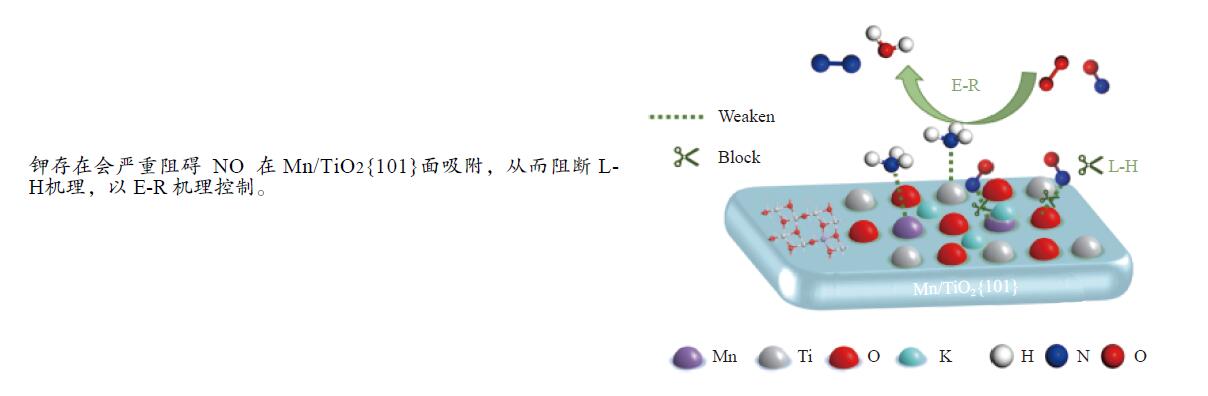
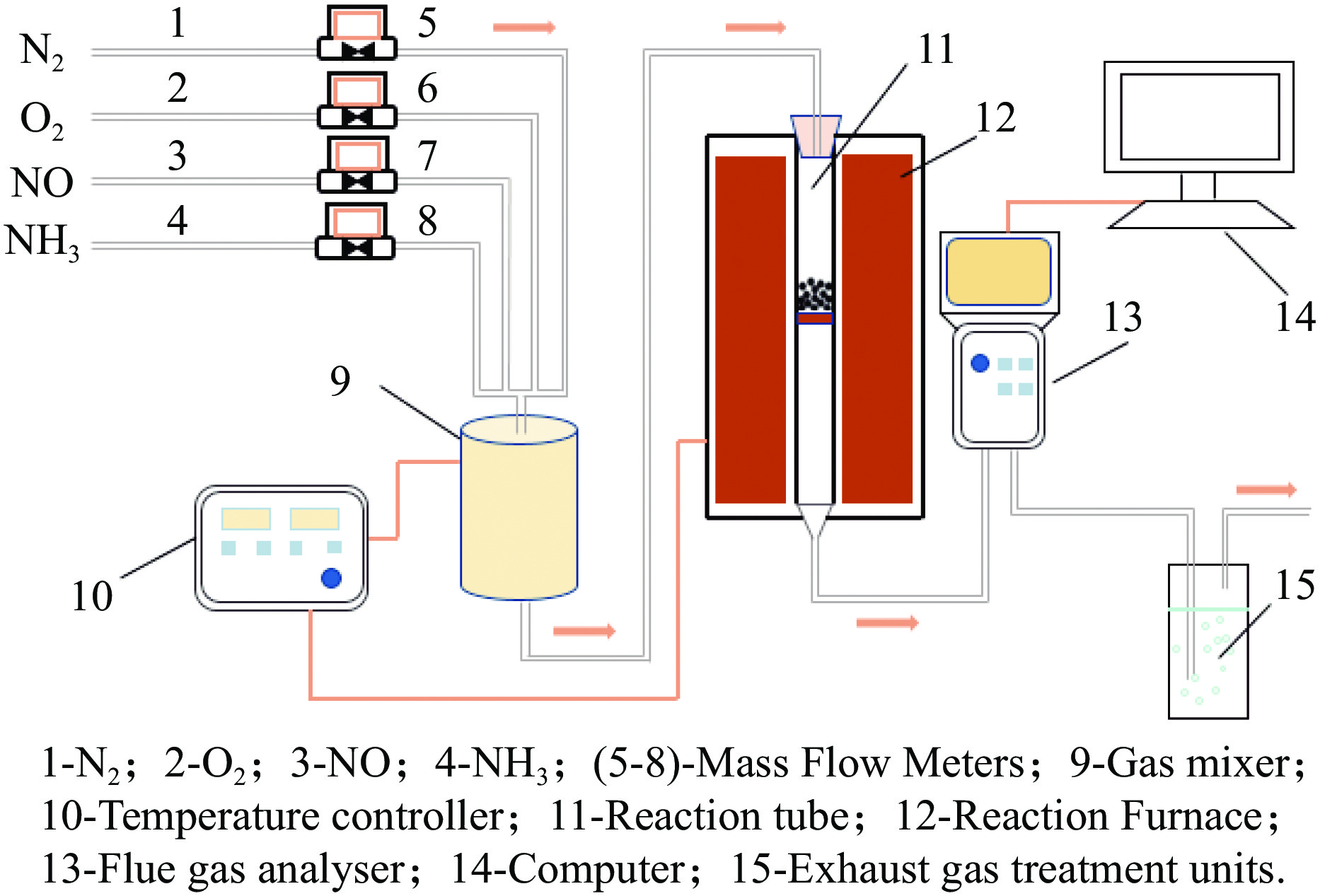
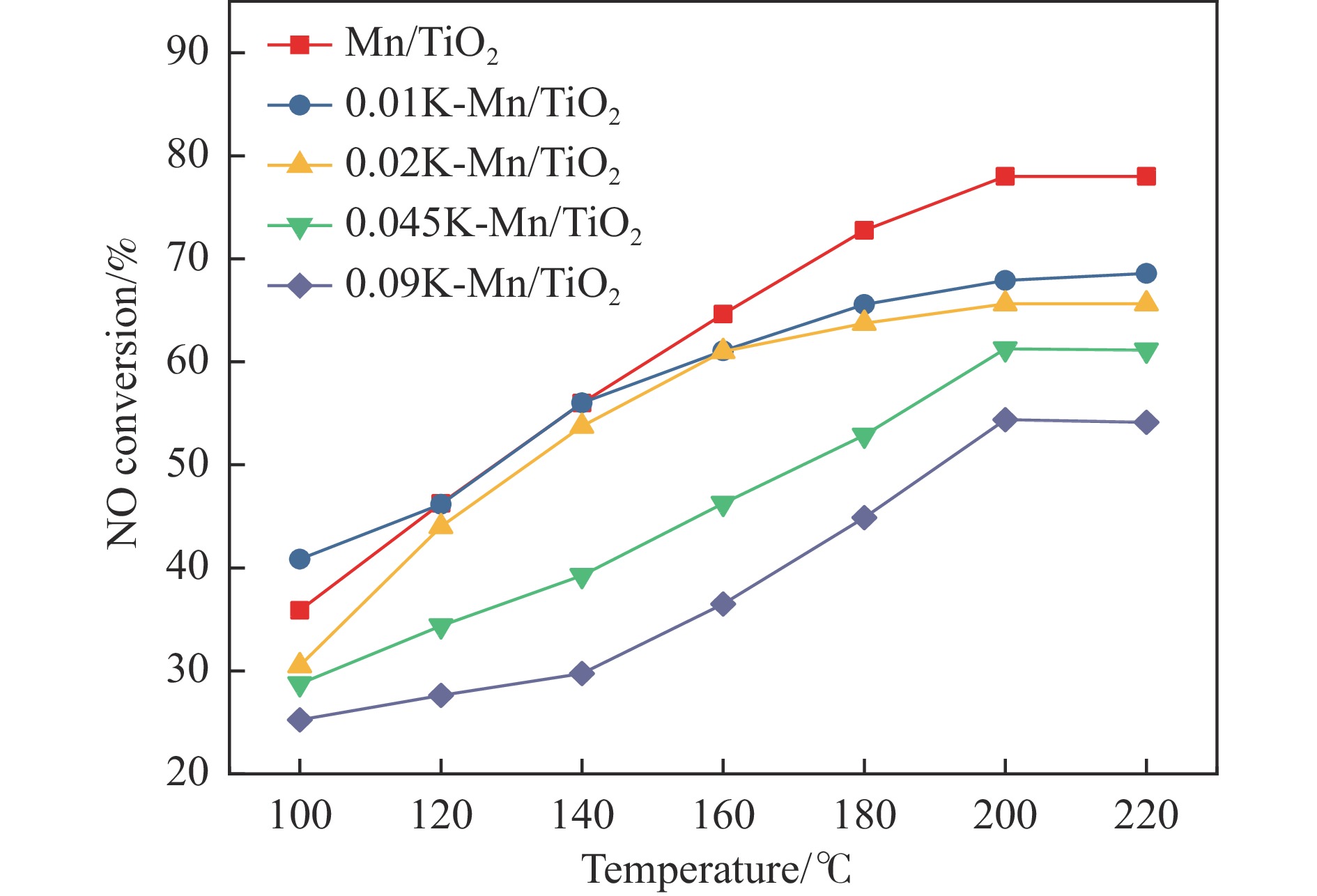
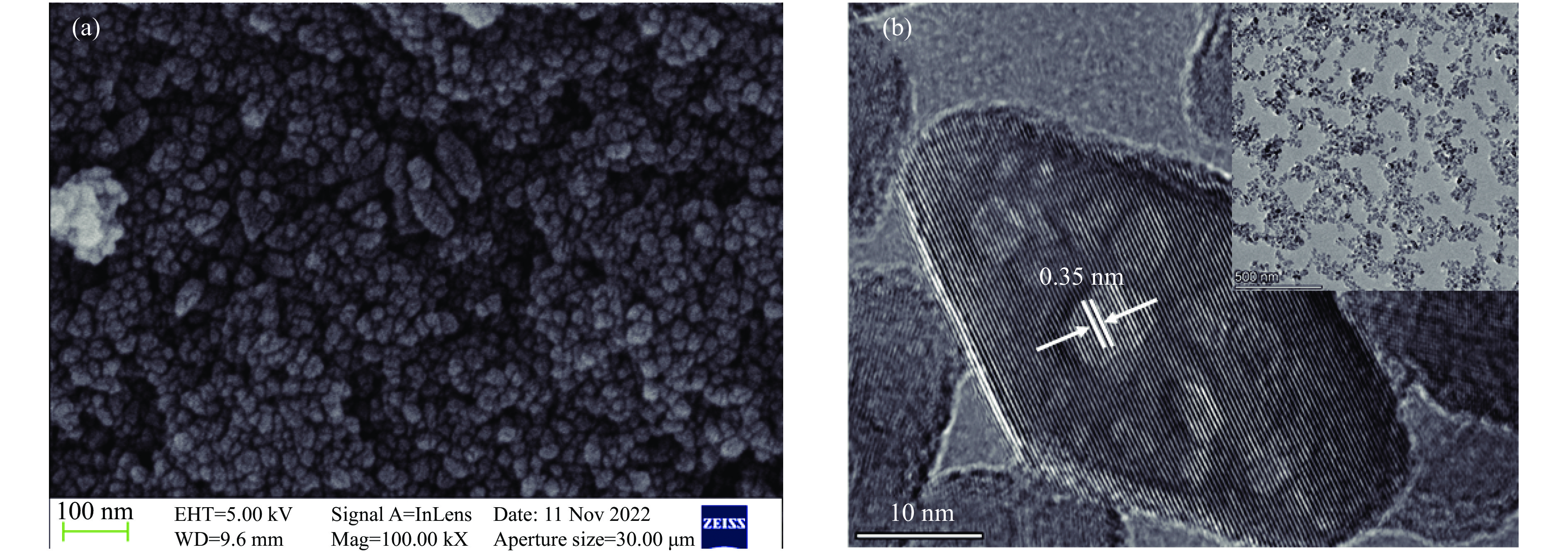
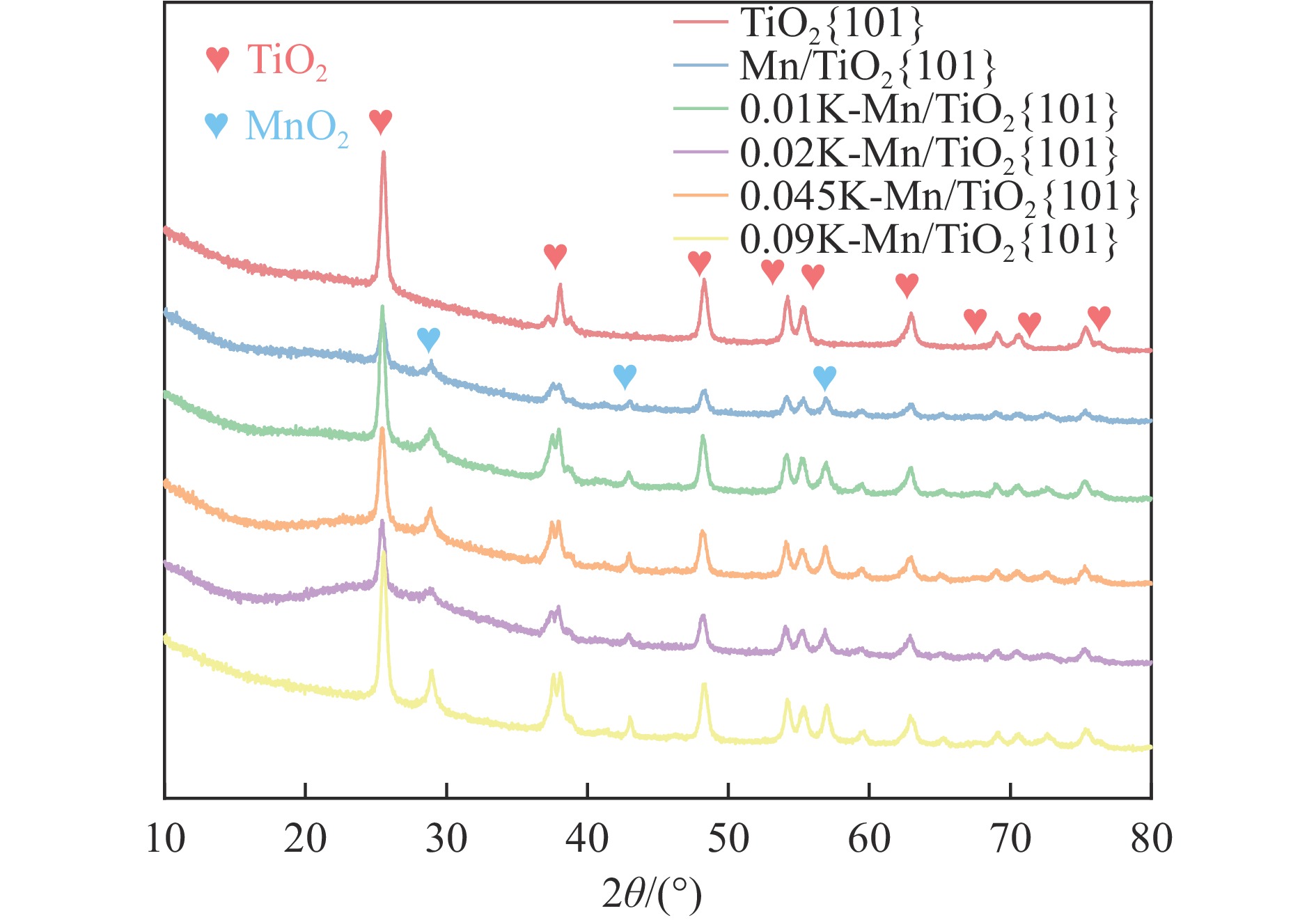
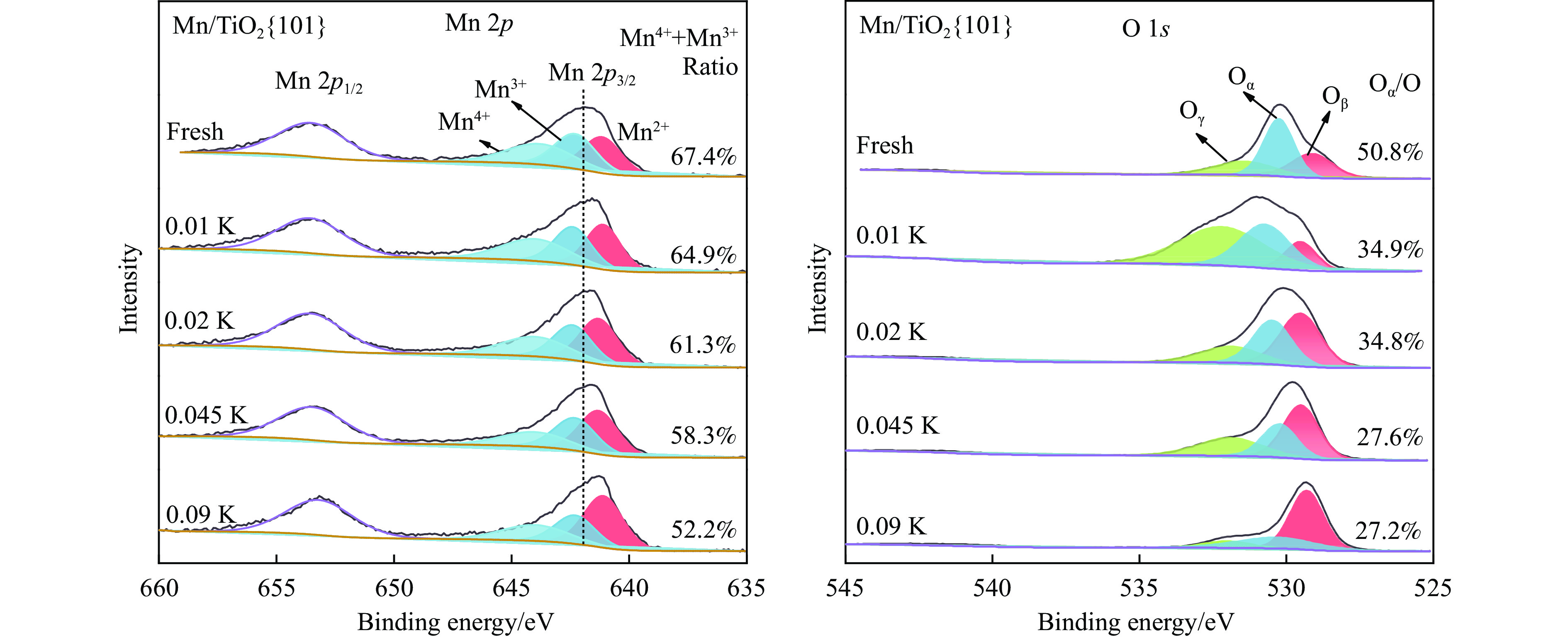
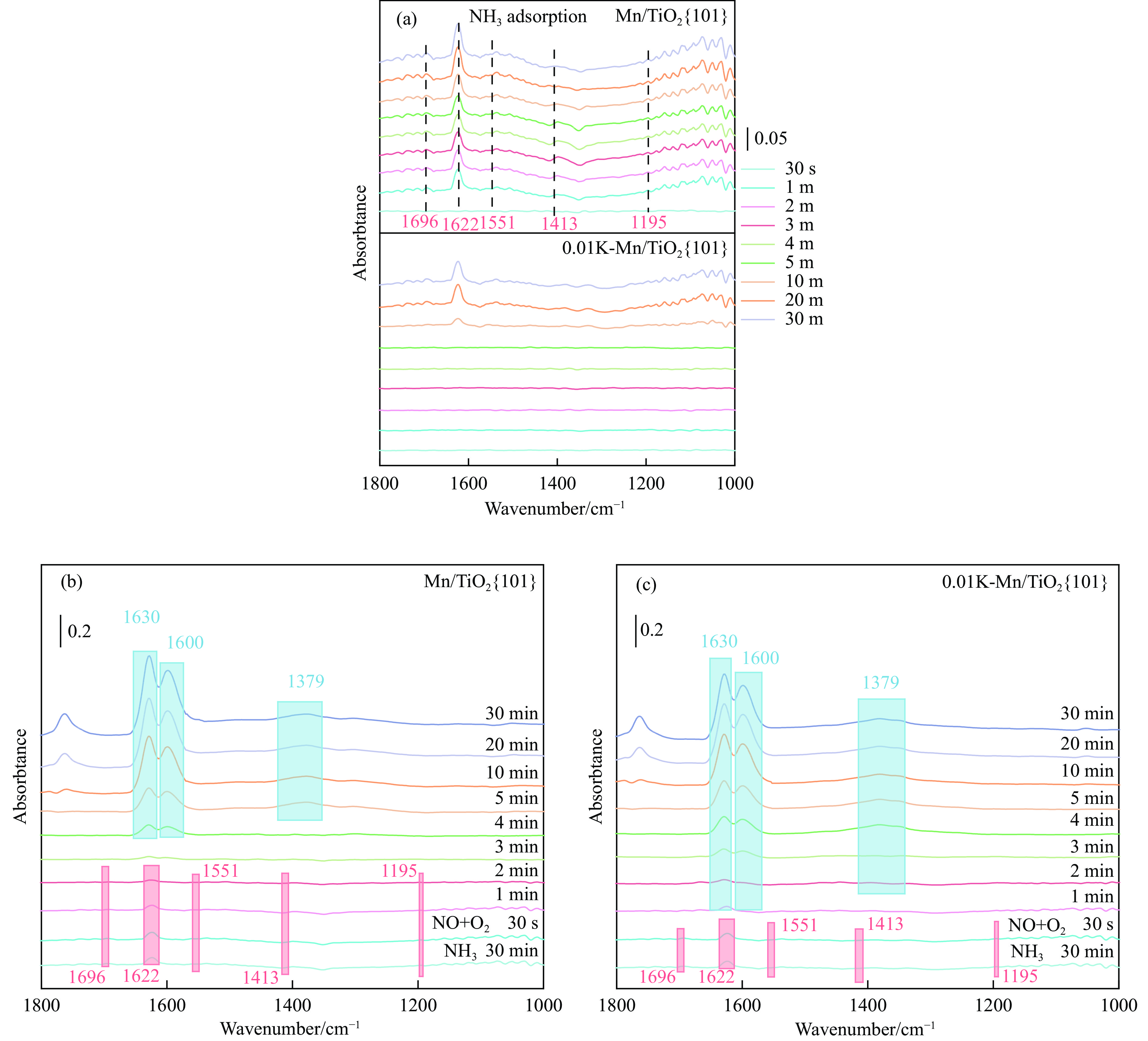
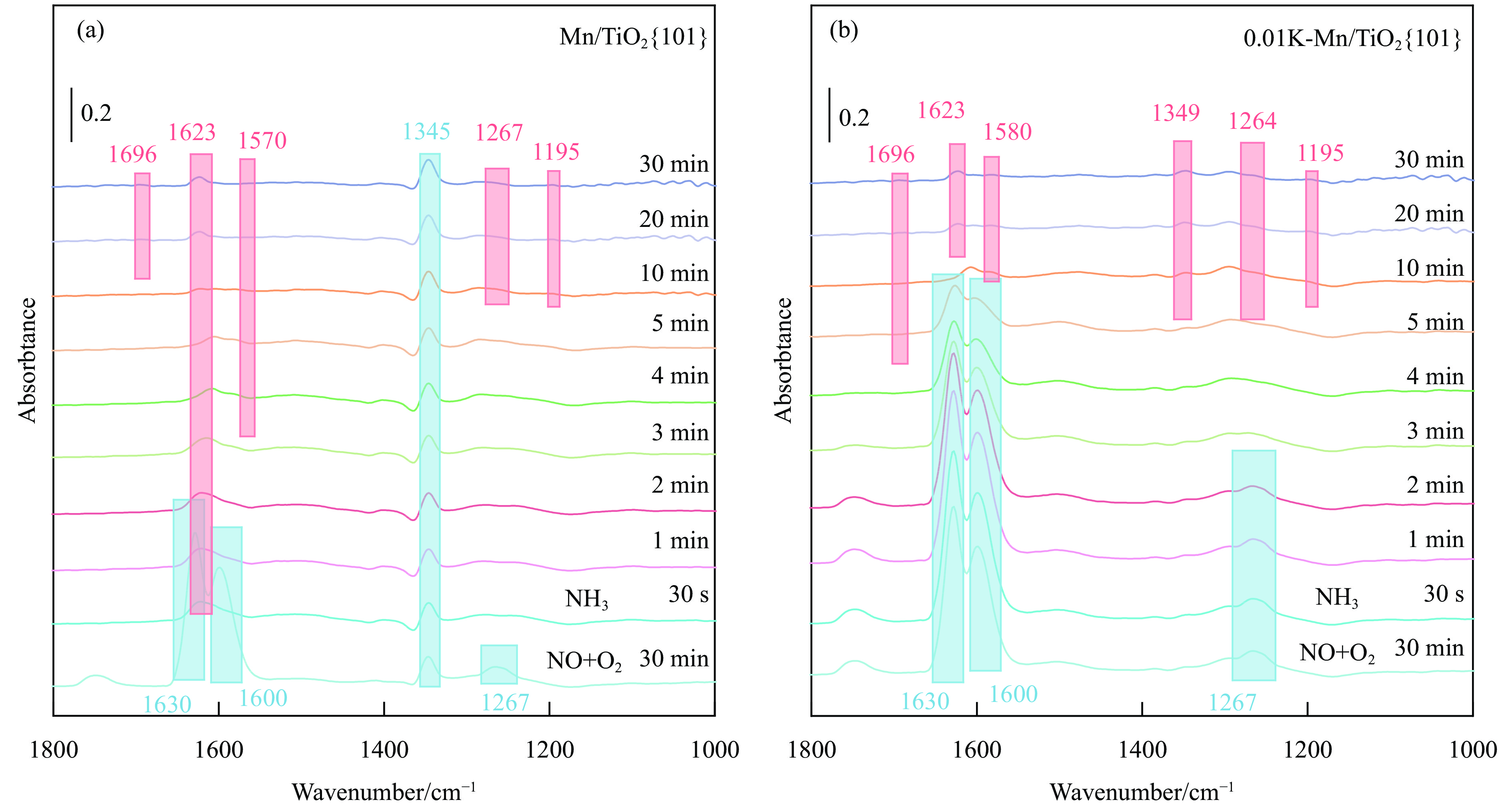
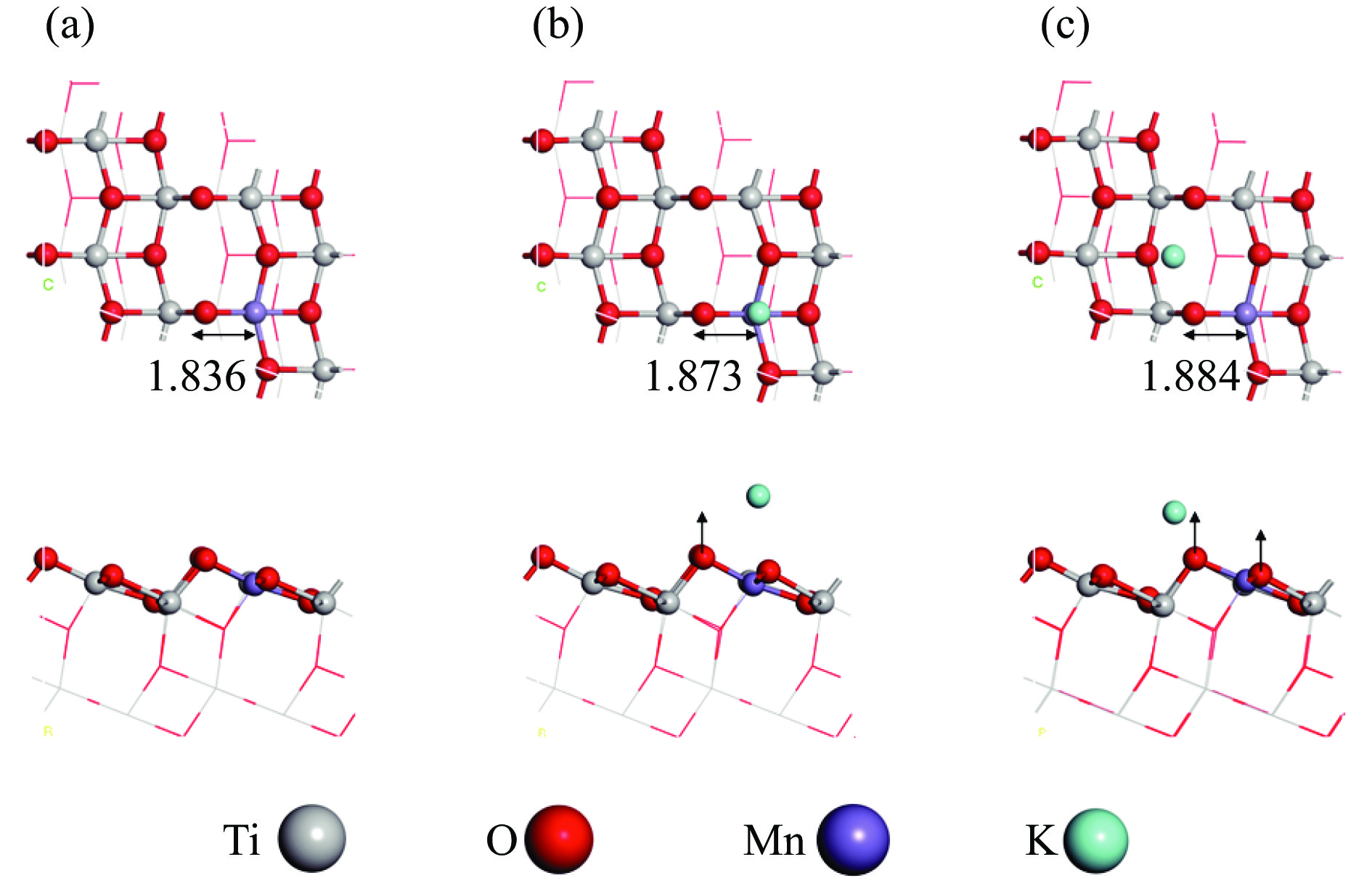
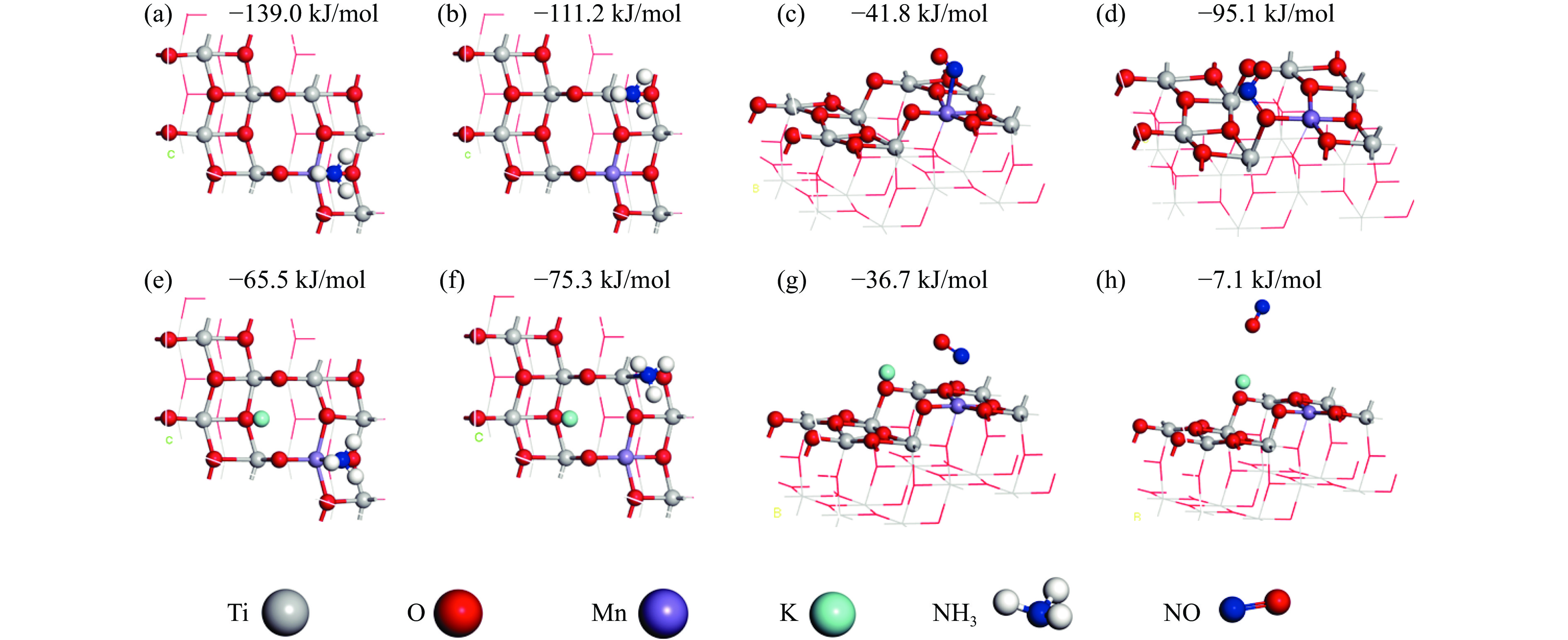
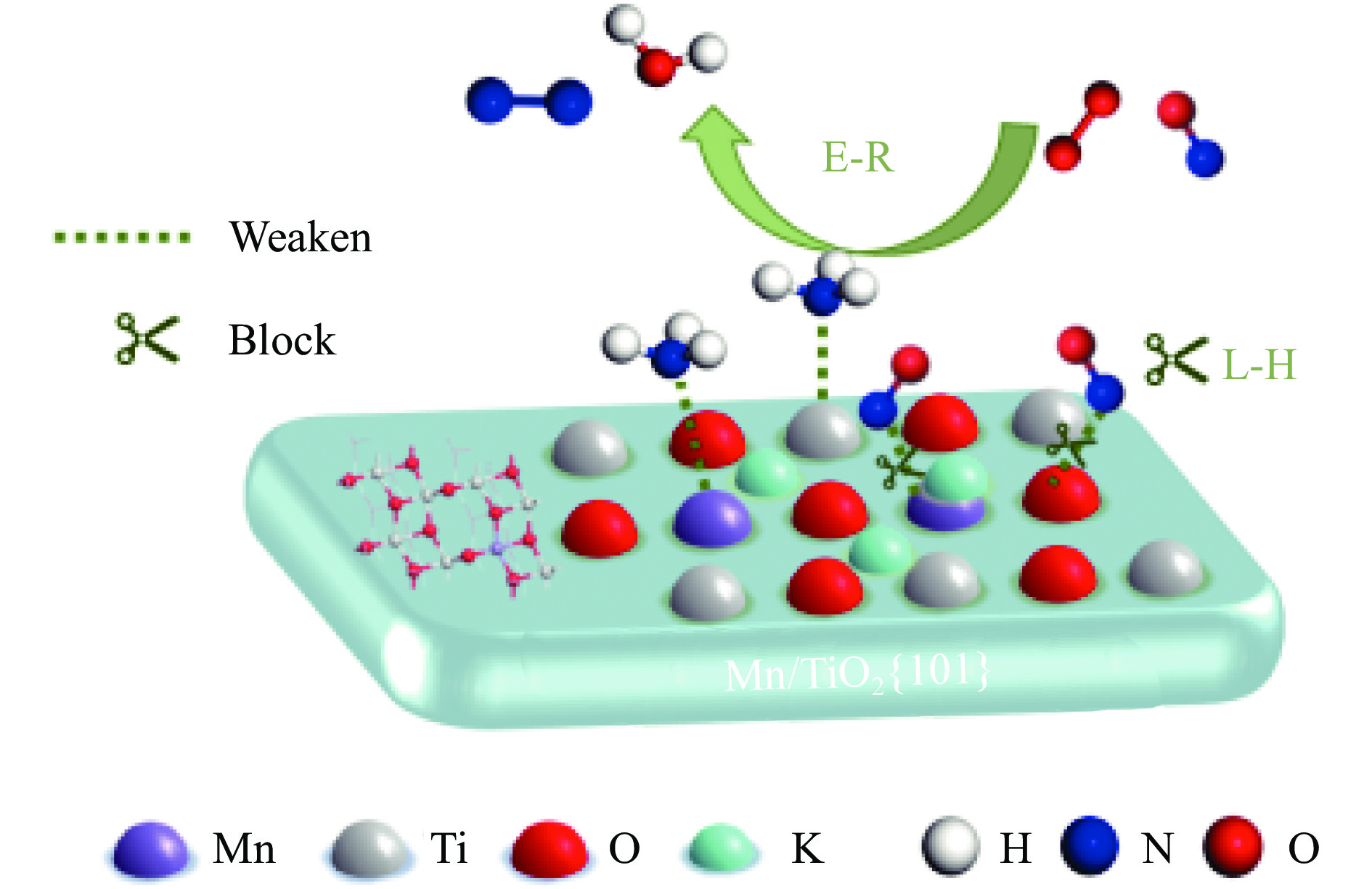
摘要: Mn/TiO2 具有良好的低温NH3 选择性催化还原NOx 2 催化剂中毒失活。论文以暴露{101}面TiO2 为载体制备Mn/TiO2 催化剂,采用浸渍法制备K中毒催化剂,研究了Mn/TiO2 低温SCR催化剂钾中毒机理。实验发现,Mn/TiO2 催化剂脱硝效率随K中毒浓度增加而减少;新鲜Mn/TiO2 催化剂表面NH3 -SCR反应由E-R和L-H机理共同控制;K吸附会导致催化剂比表面积降低,催化剂表面Mn4+ 、化学吸附氧比例降低,表面酸性位点数量减少,导致脱硝活性降低;同时K更易吸附在Mn顶位以及桥接O位附近,导致NO的吸附活化受到严重遏制,同时削弱NH3 的吸附,使得L-H机理受到阻断,只能以E-R机理控制为主。
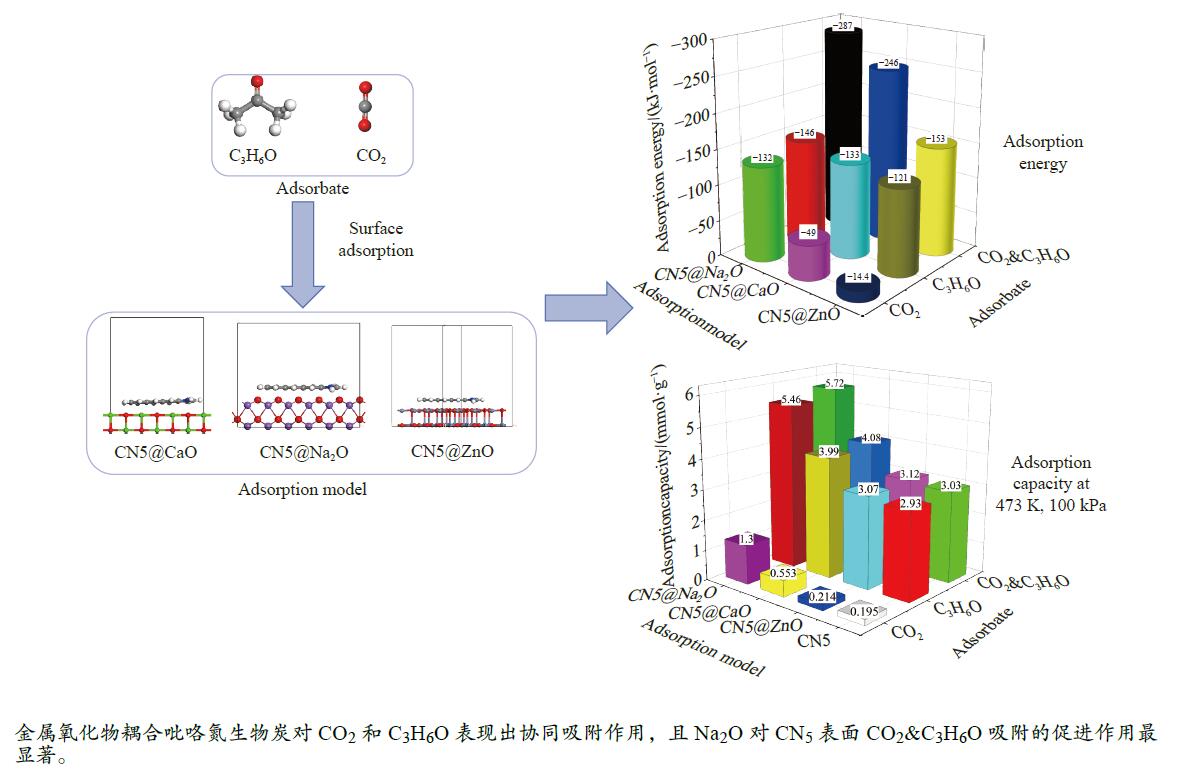
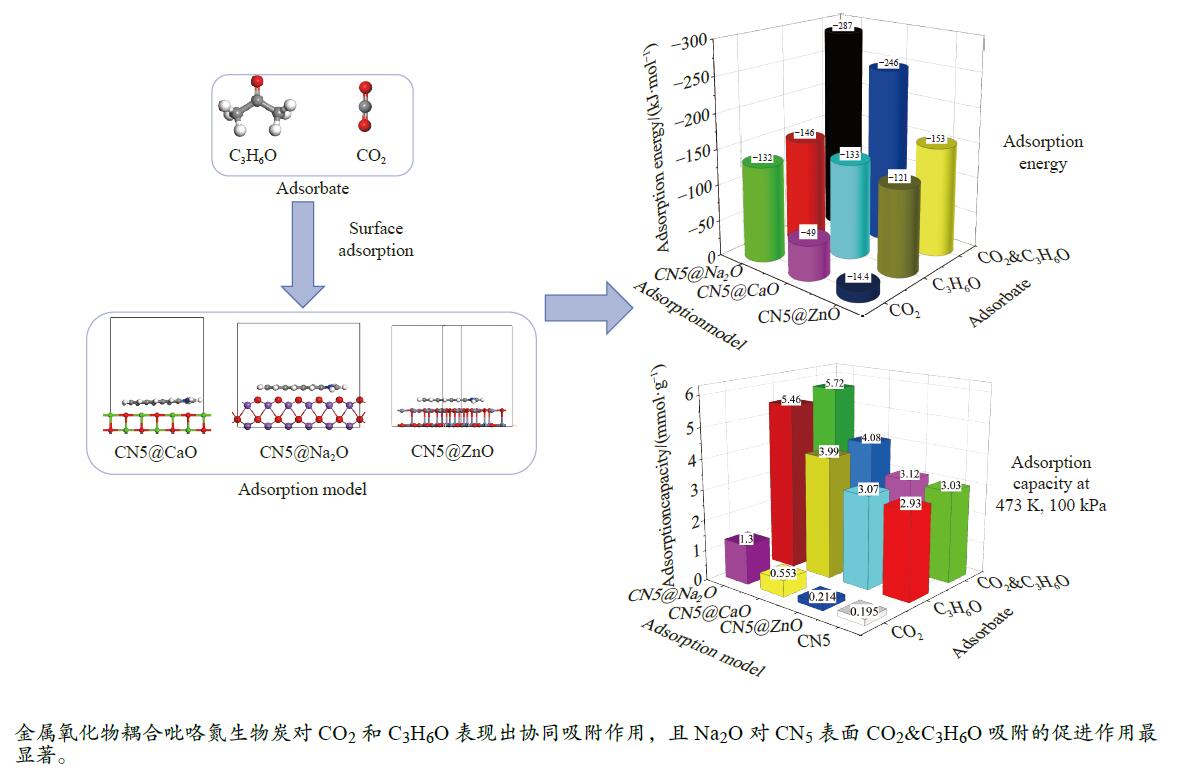
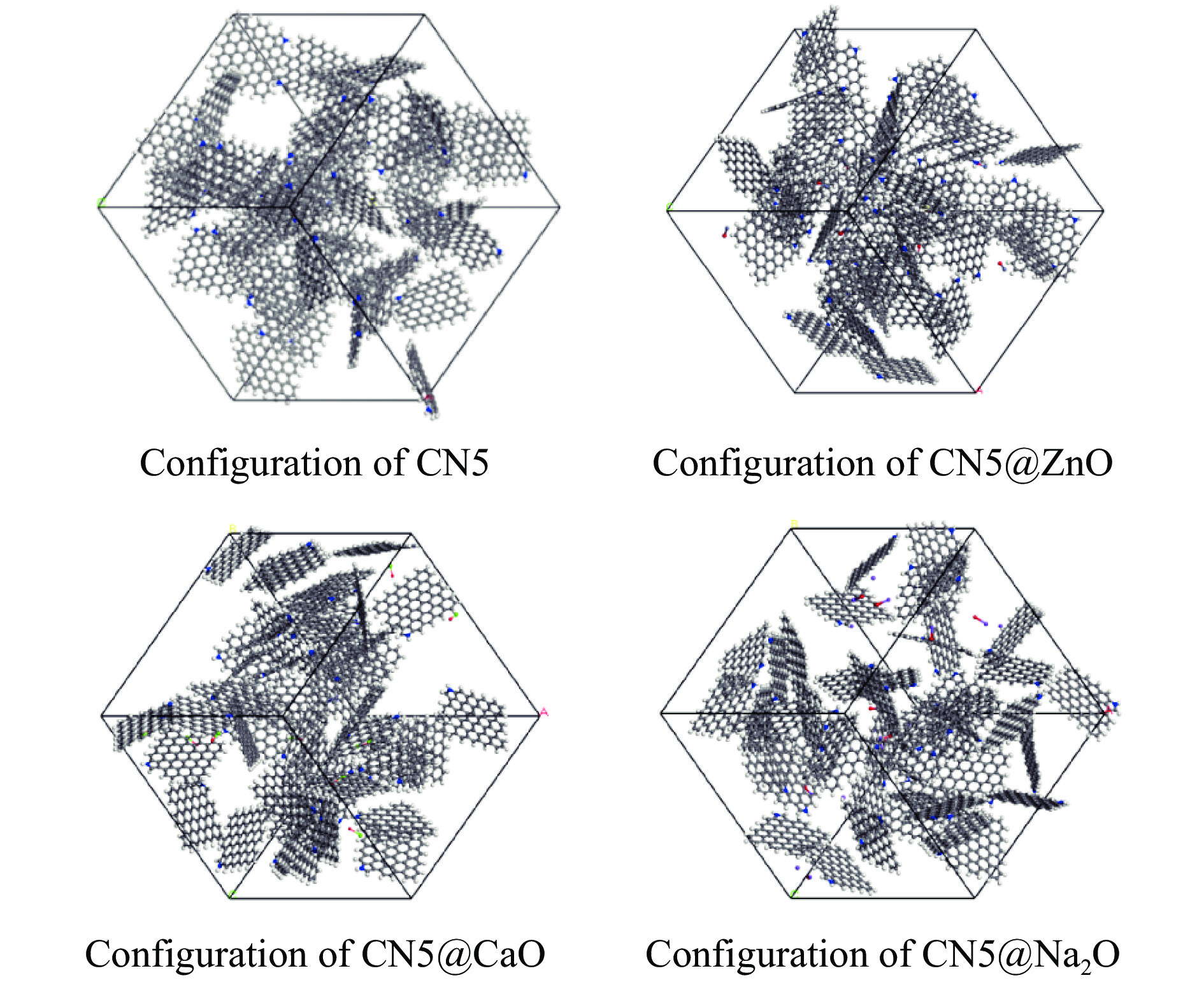
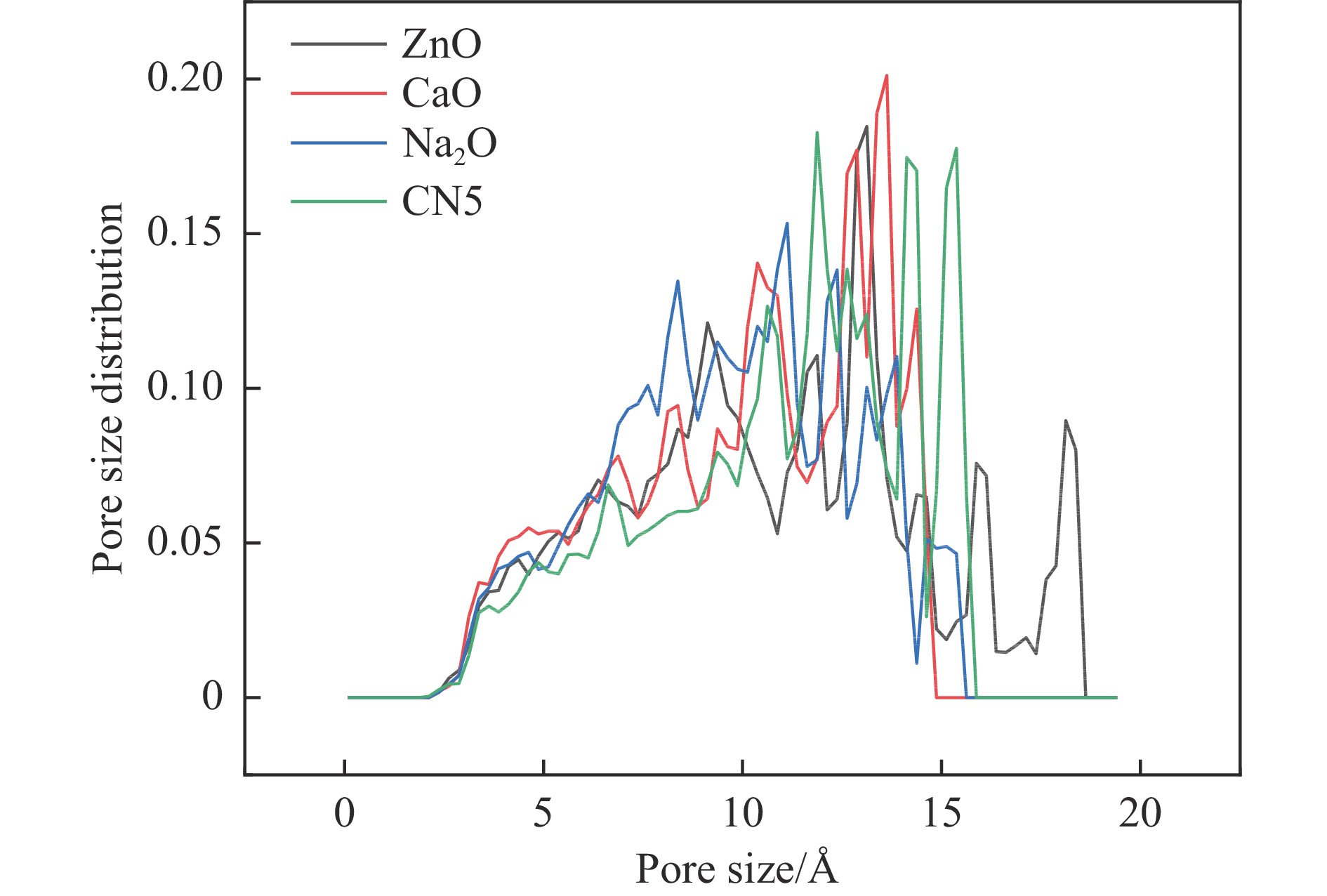
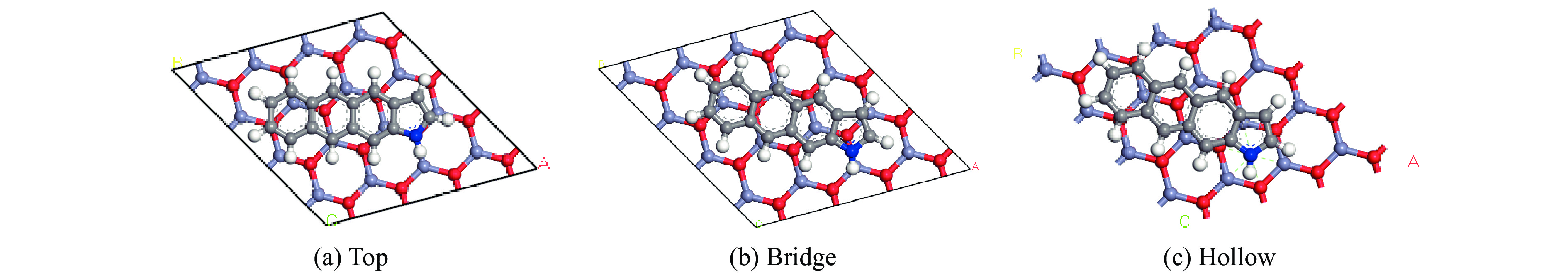
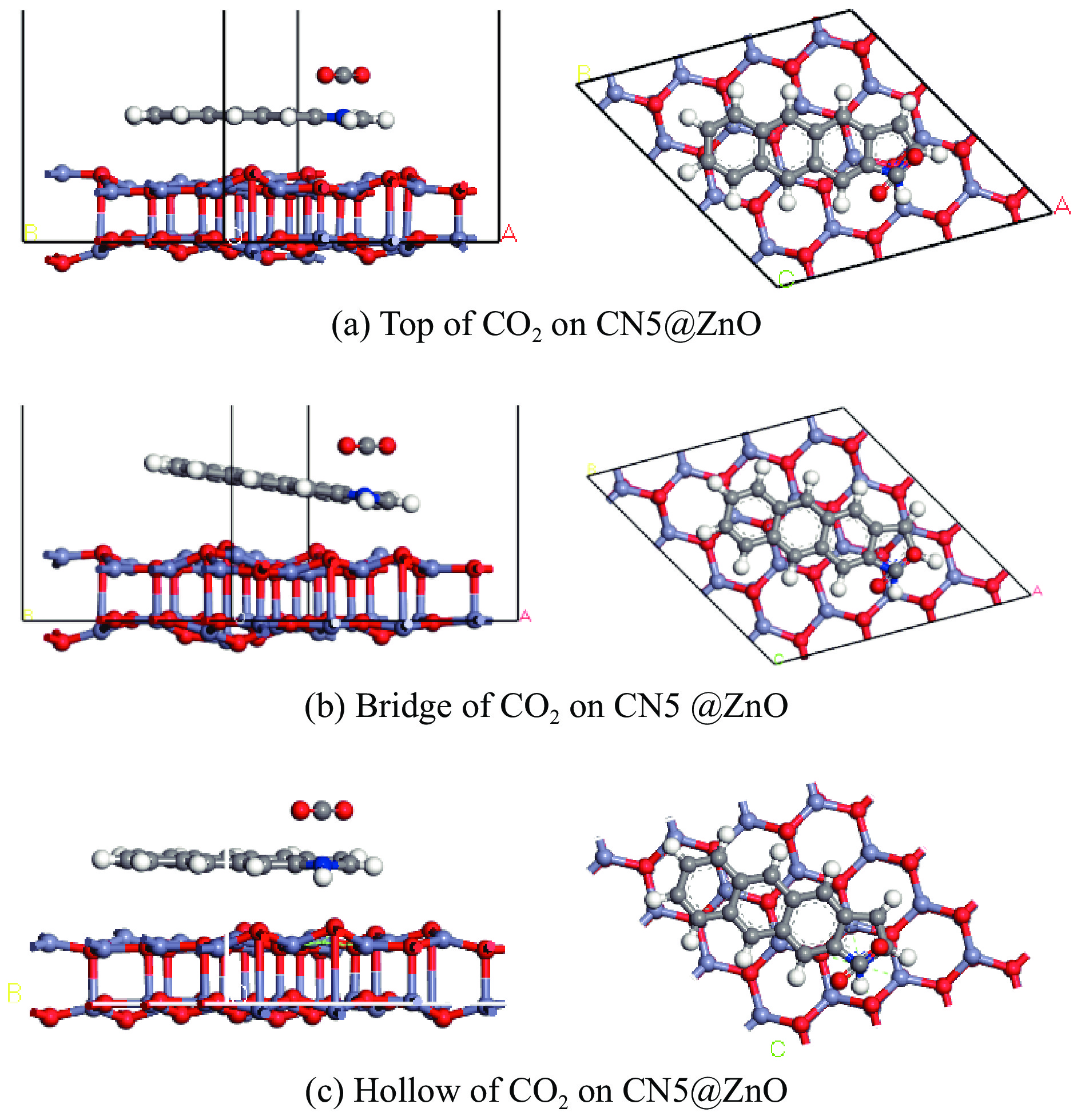
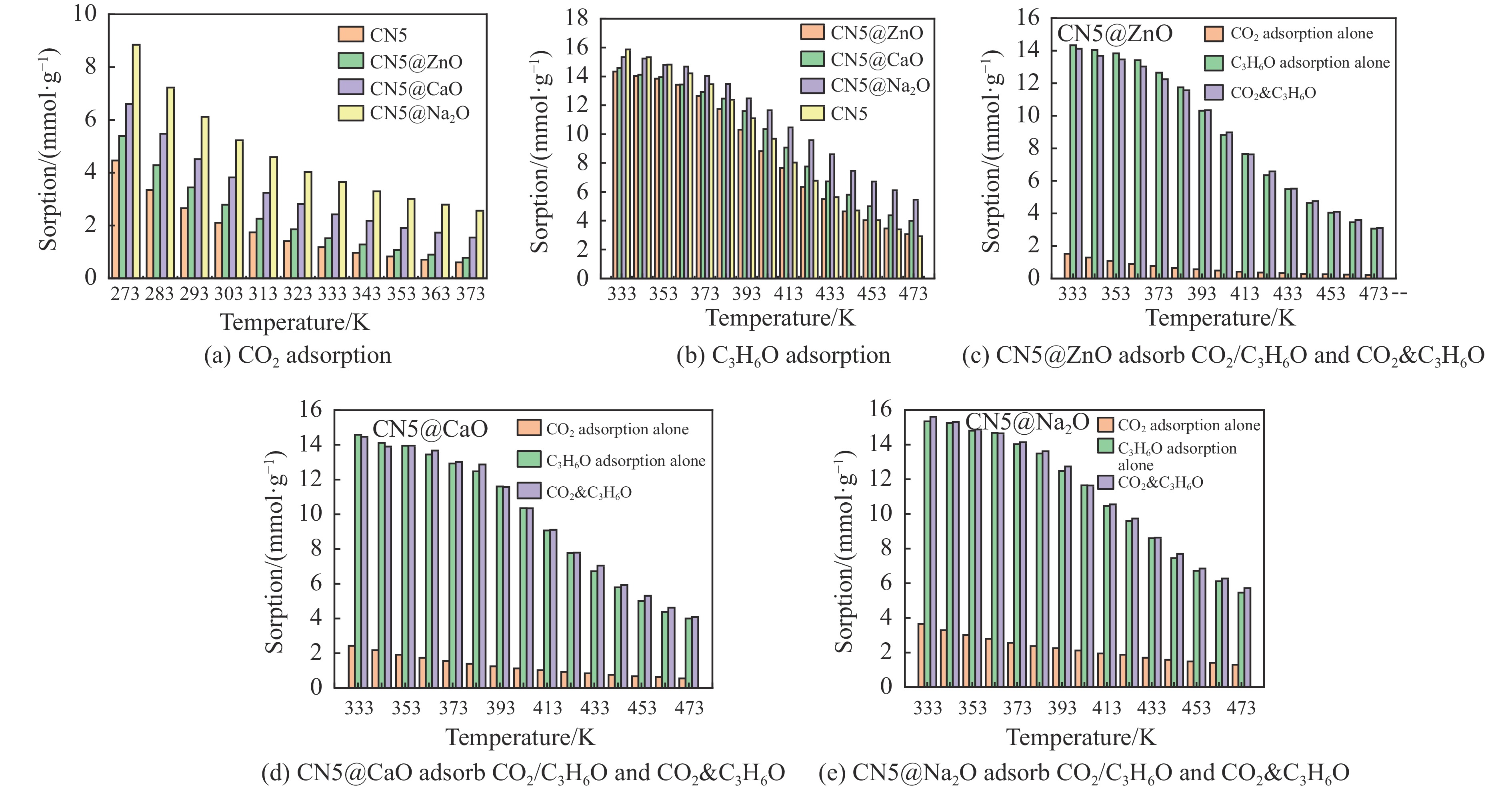
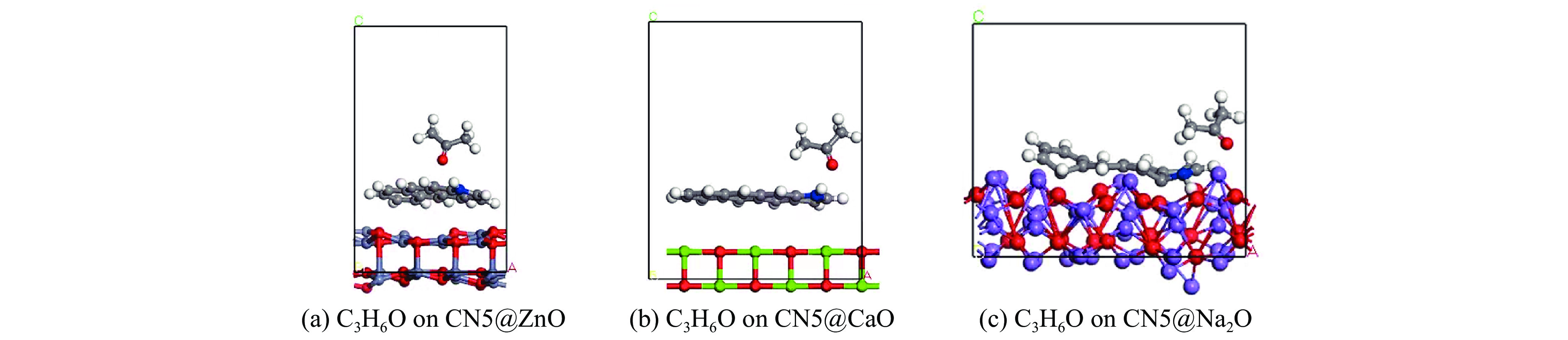
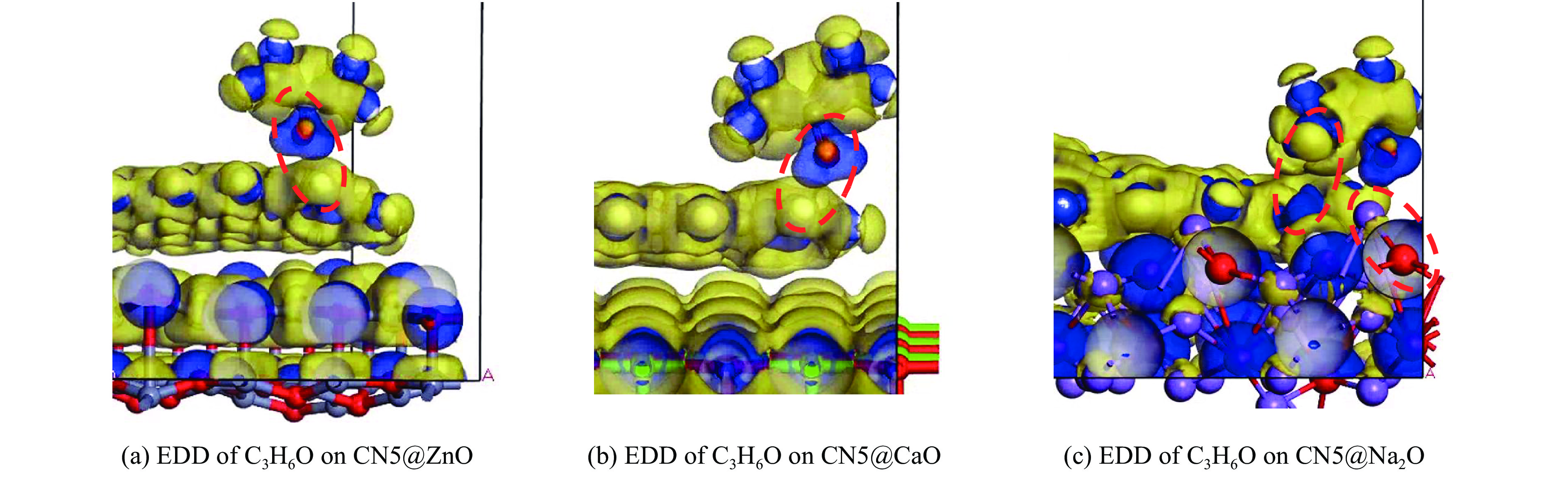
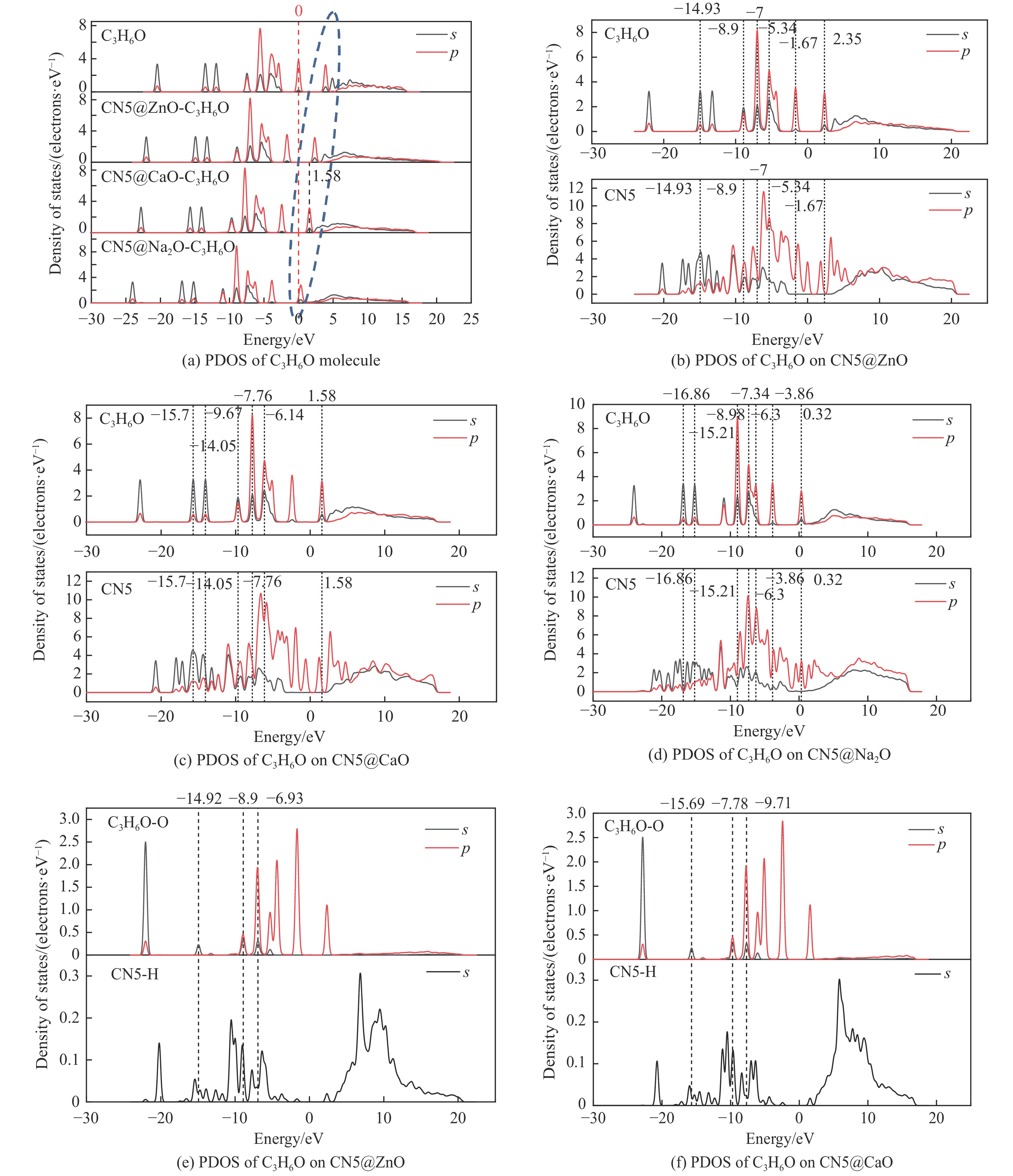
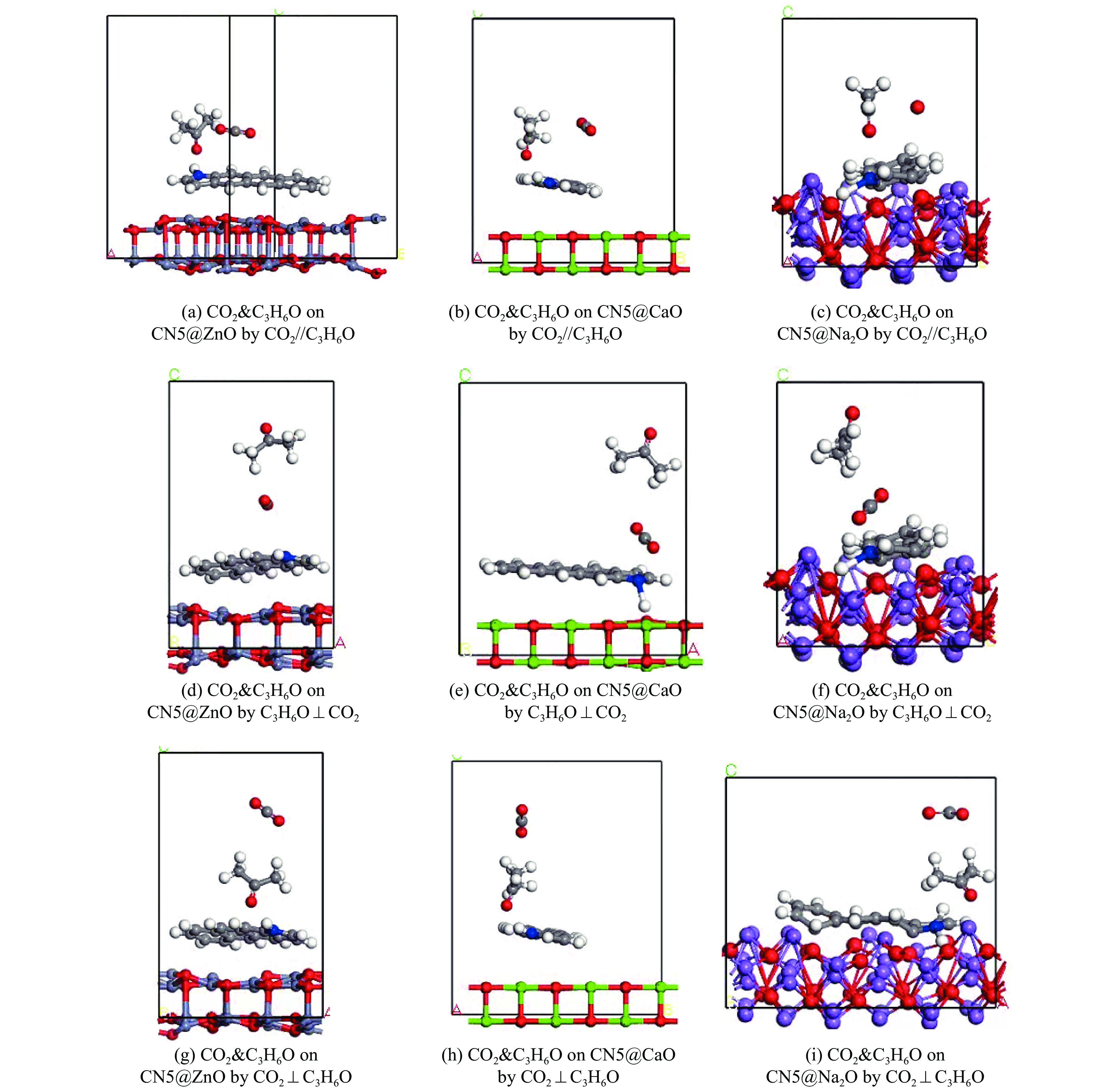
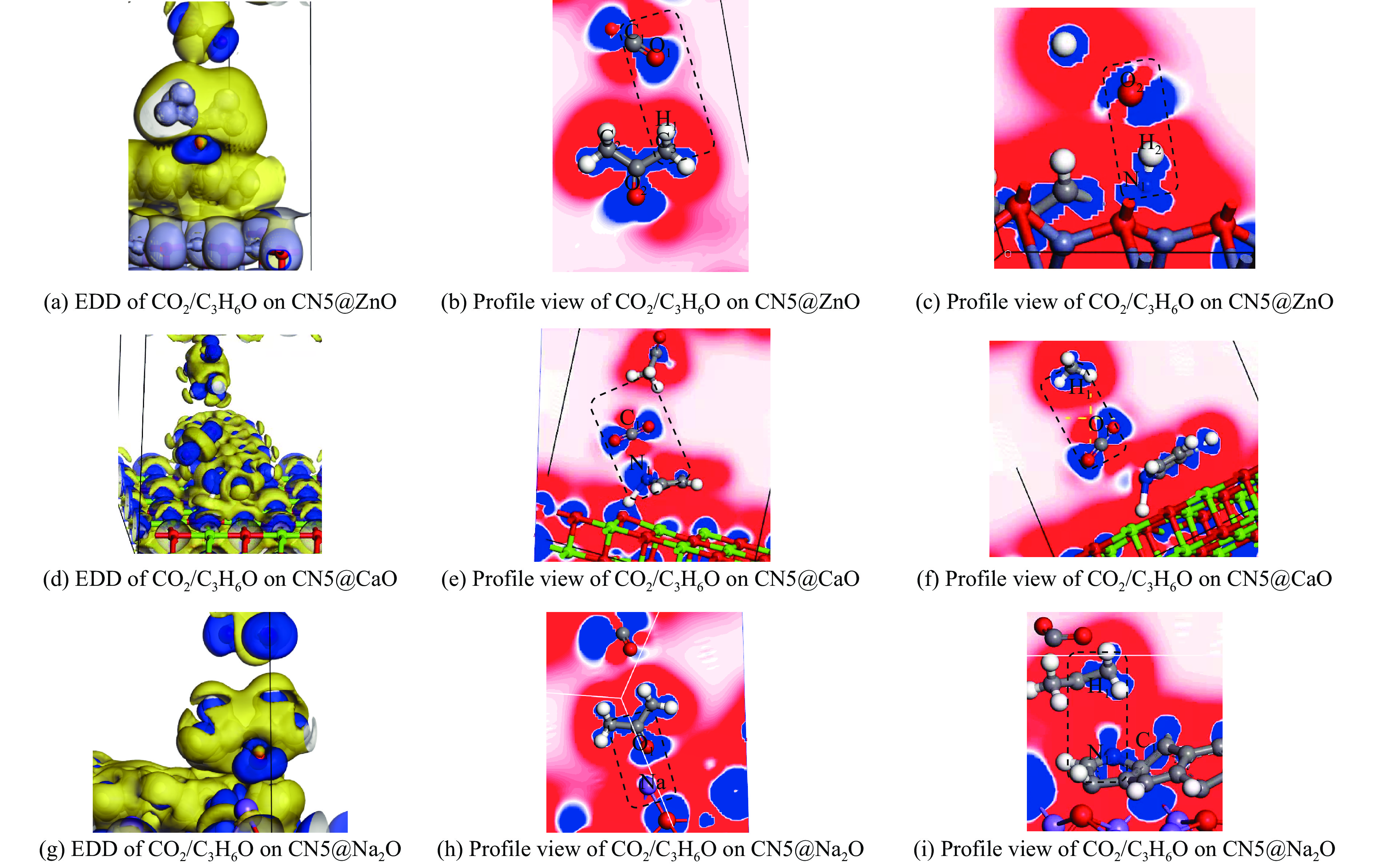
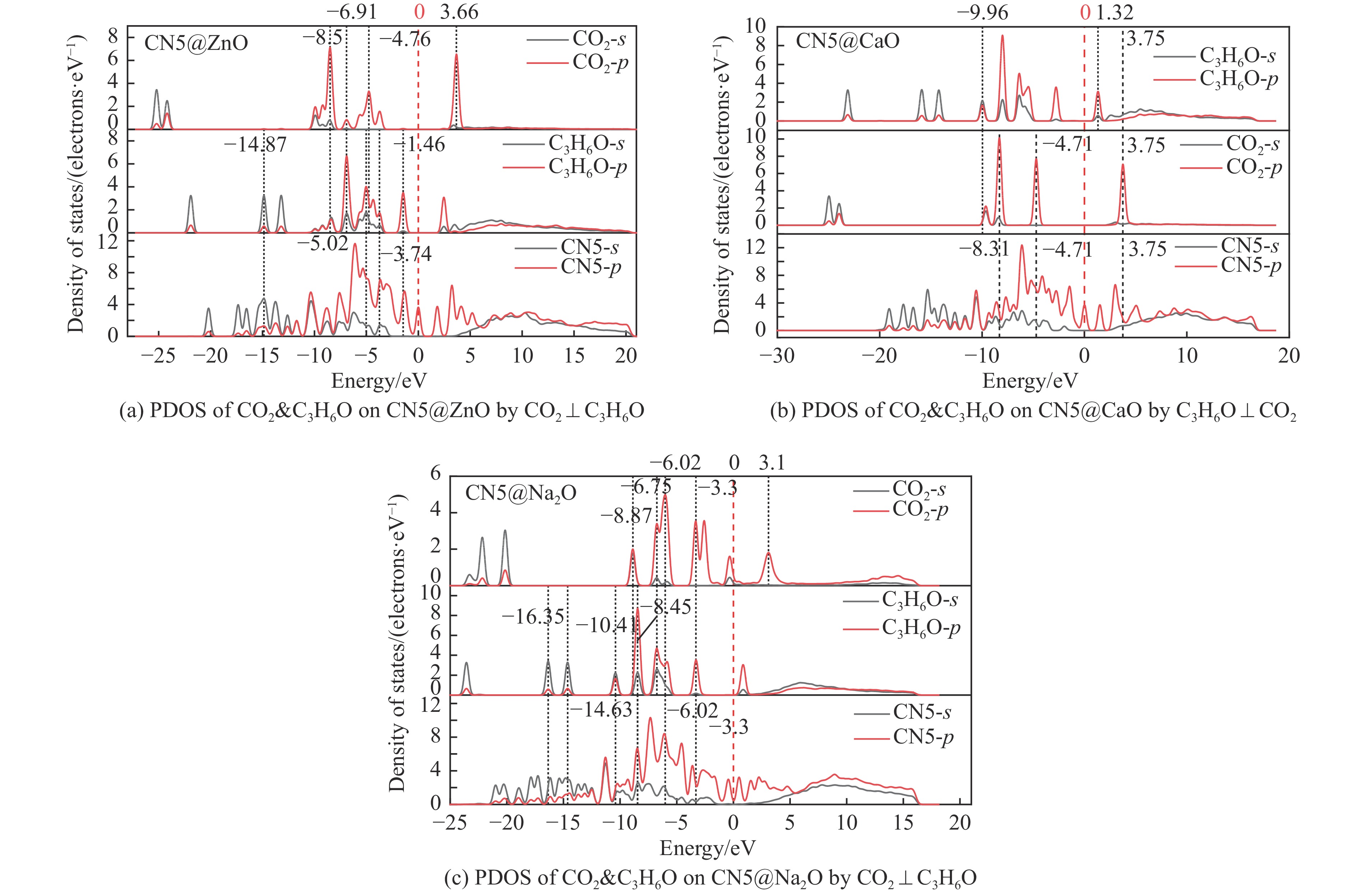
摘要: 本研究采用密度泛函理论,通过比较吸附量、吸附能以及态密度和电荷差分密度的分析,探究了不同金属氧化物耦合吡咯氮生物炭(CN5@MOx x 2 O)表面CO2 与C3 H6 O(CO2 &C3 H6 O)的吸附机理。首先从CO2 /C3 H6 O单组分方面计算了其在CN5@MOx 2 O表面对CO2 /C3 H6 O单组分吸附量分别为3.65、15.34 mmol/g,吸附能分别为−145.86、−132.47 kJ/mol,均高于CO2 /C3 H6 O单组分在CN5@CaO及CN5@ZnO表面吸附。得出Na2 O掺杂吡咯氮生物炭对CO2 /C3 H6 O单组分吸附效果最优。进一步研究了CO2 &C3 H6 O在CN5@MOx 2 &C3 H6 O在CN5@Na2 O、CN5@CaO、CN5@ZnO表面吸附存在临界温度(分别为333、353、393 K),超过临界温度以后CO2 &C3 H6 O共存体系在CN5@MOx 2 /C3 H6 O单组分有所提高。CO2 &C3 H6 O在CN5@Na2 O、CN5@CaO、CN5@ZnO表面吸附能分别比CO2 或C3 H6 O单组分吸附时至少高141.59、112.77、31.75 kJ/mol,CN5@MOx 2 和C3 H6 O的吸附表现为协同促进作用,且CN5@Na2 O对CO2 &C3 H6 O共同吸附效果最佳。采用电荷差分密度和态密度分析CO2 &C3 H6 O在CN5@MOx 2 的吸附作用力是通过C3 H6 O与CO2 的间接相互作用产生的,Na2 O中Na与C3 H6 O电子云重叠,发生电荷转移,增强了两者间相互作用力,CN5@Na2 O表面C3 H6 O与CN5在p 轨道主要共振峰结合能较CN5@ZnO低了3.43 eV,使得C3 H6 O在CN5@Na2 O表面吸附最稳定。
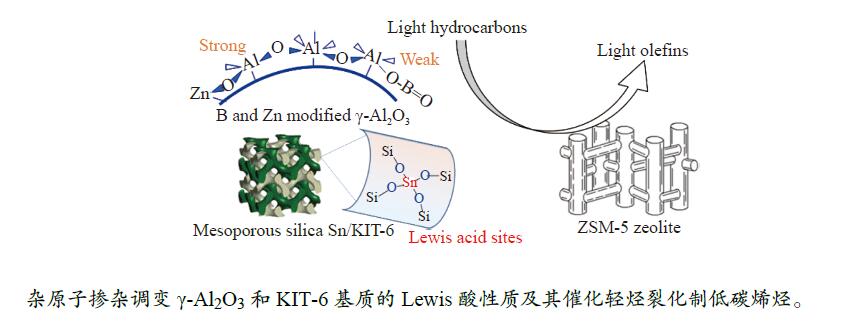
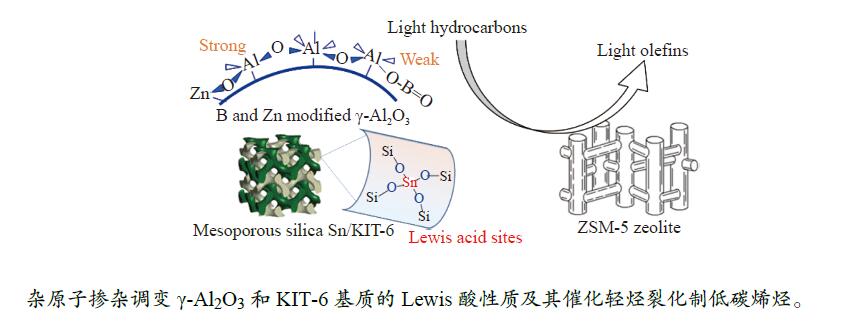
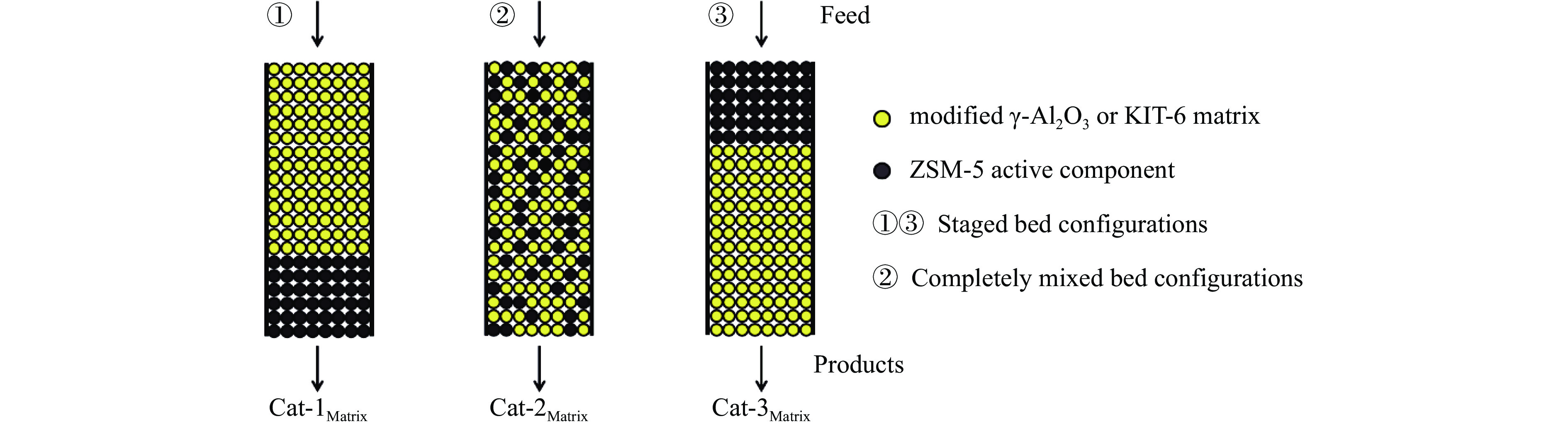
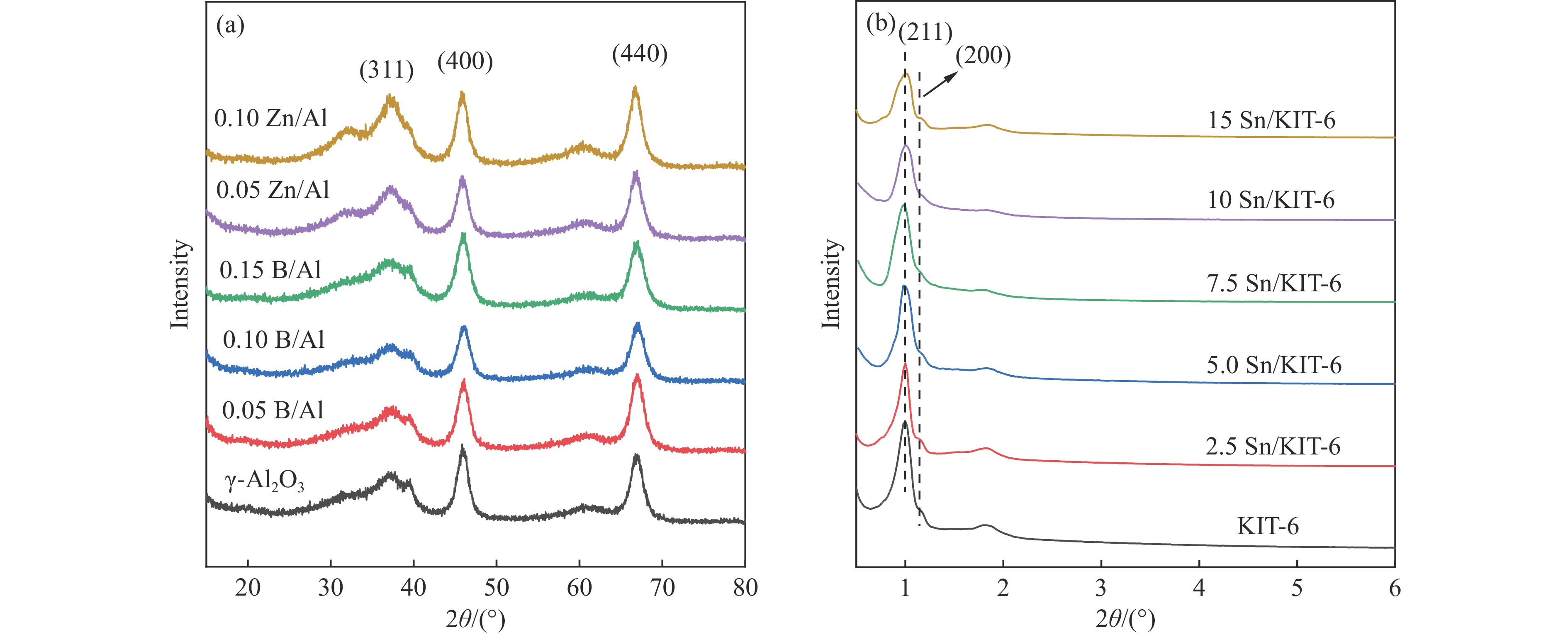
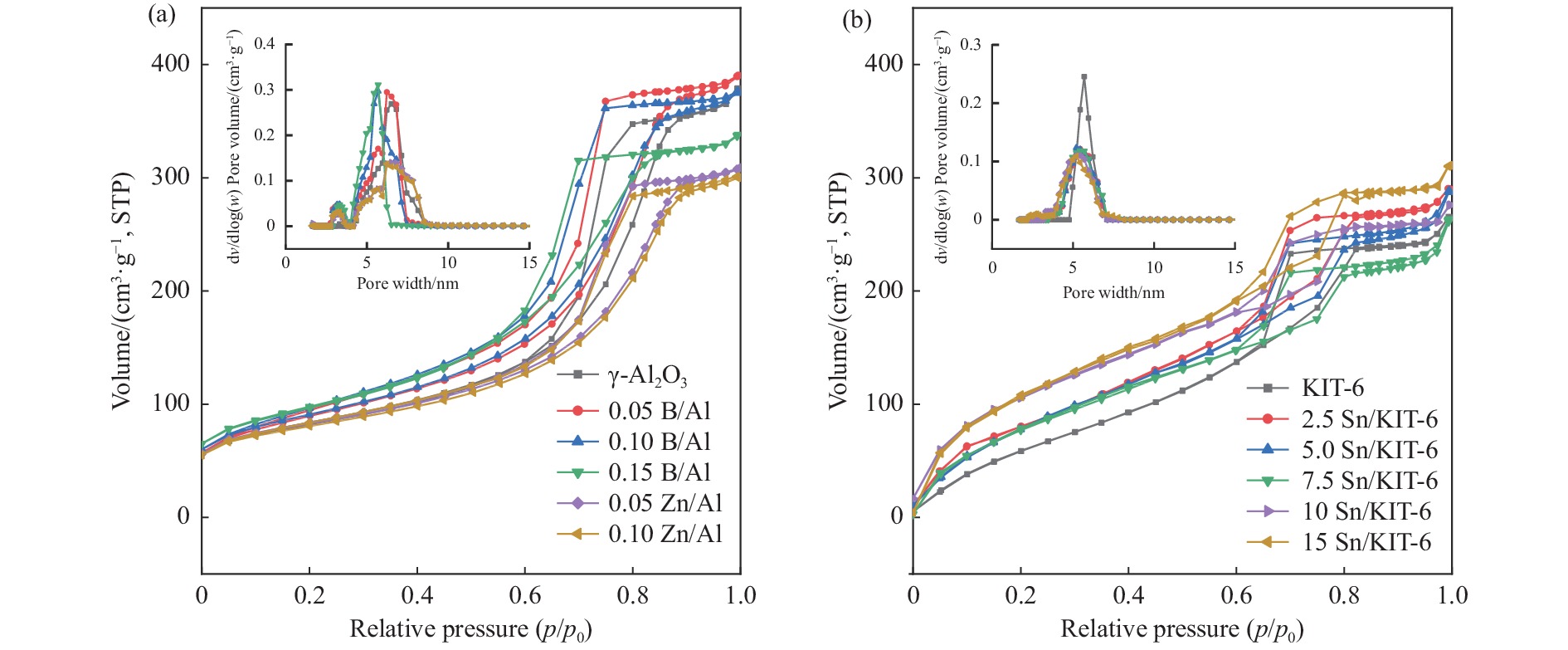
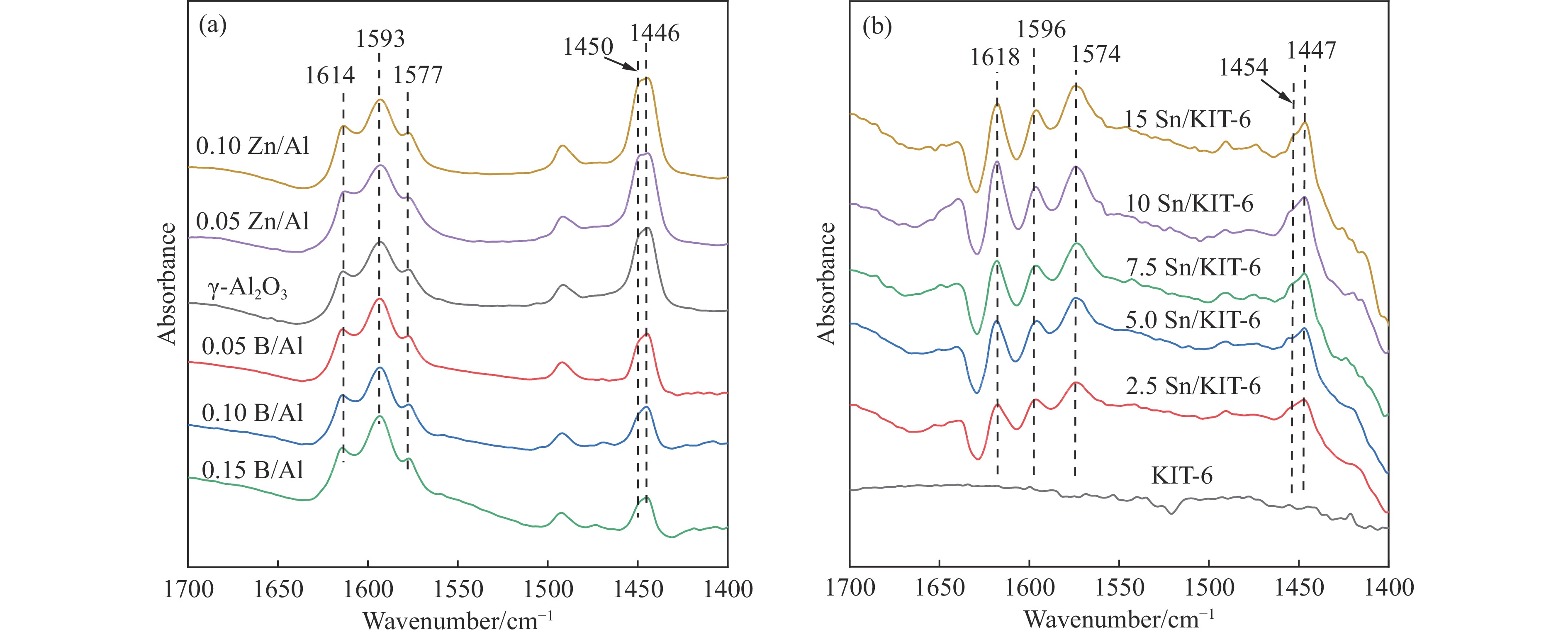
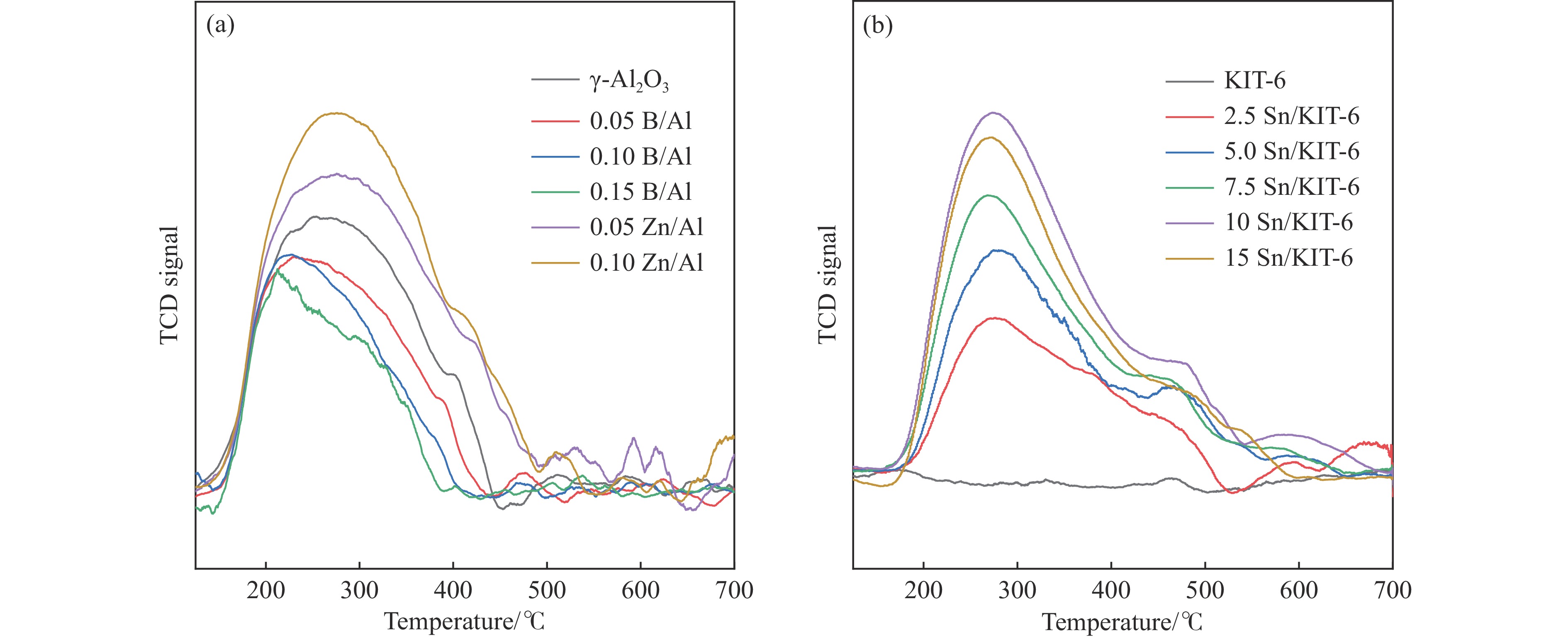
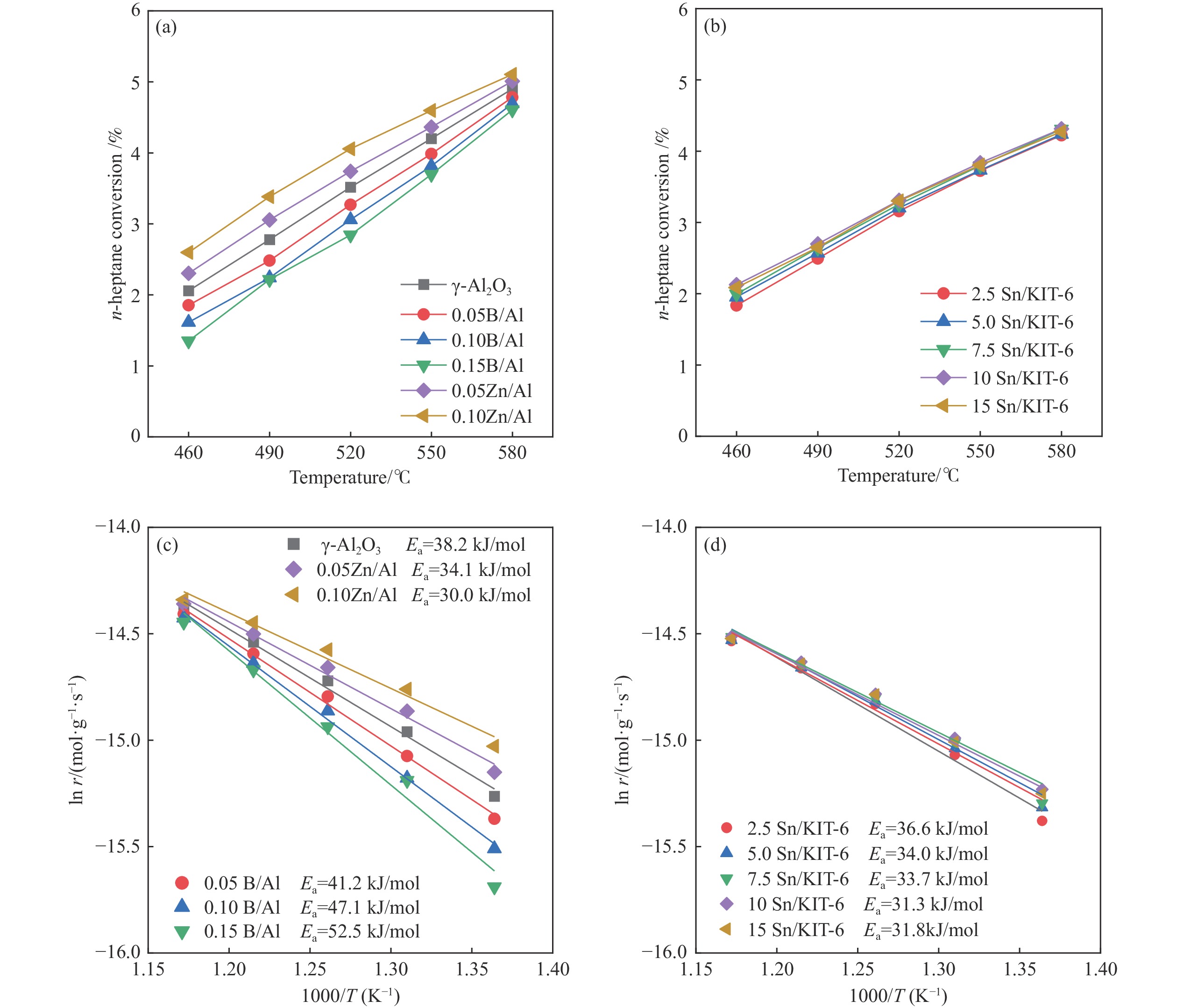
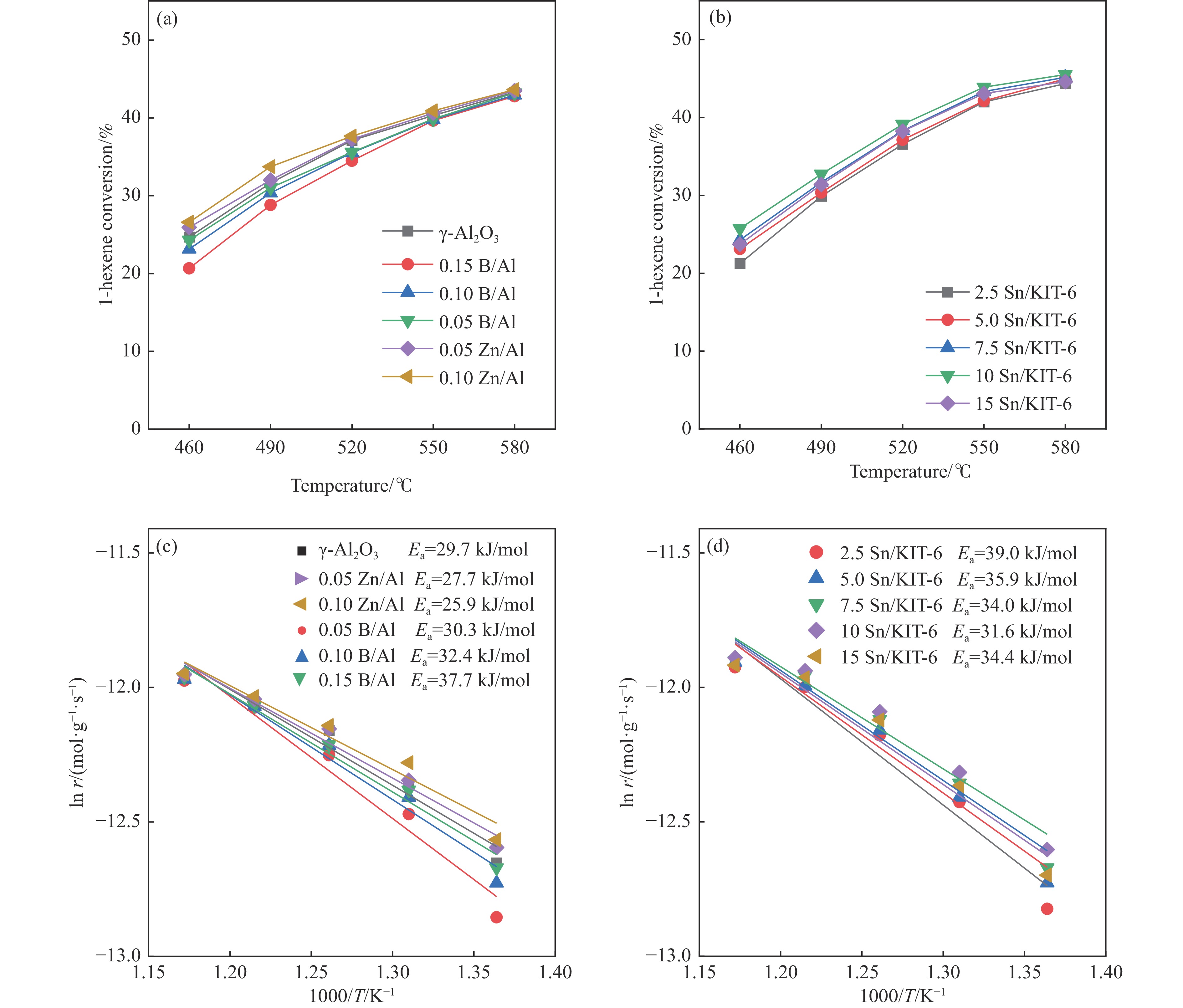
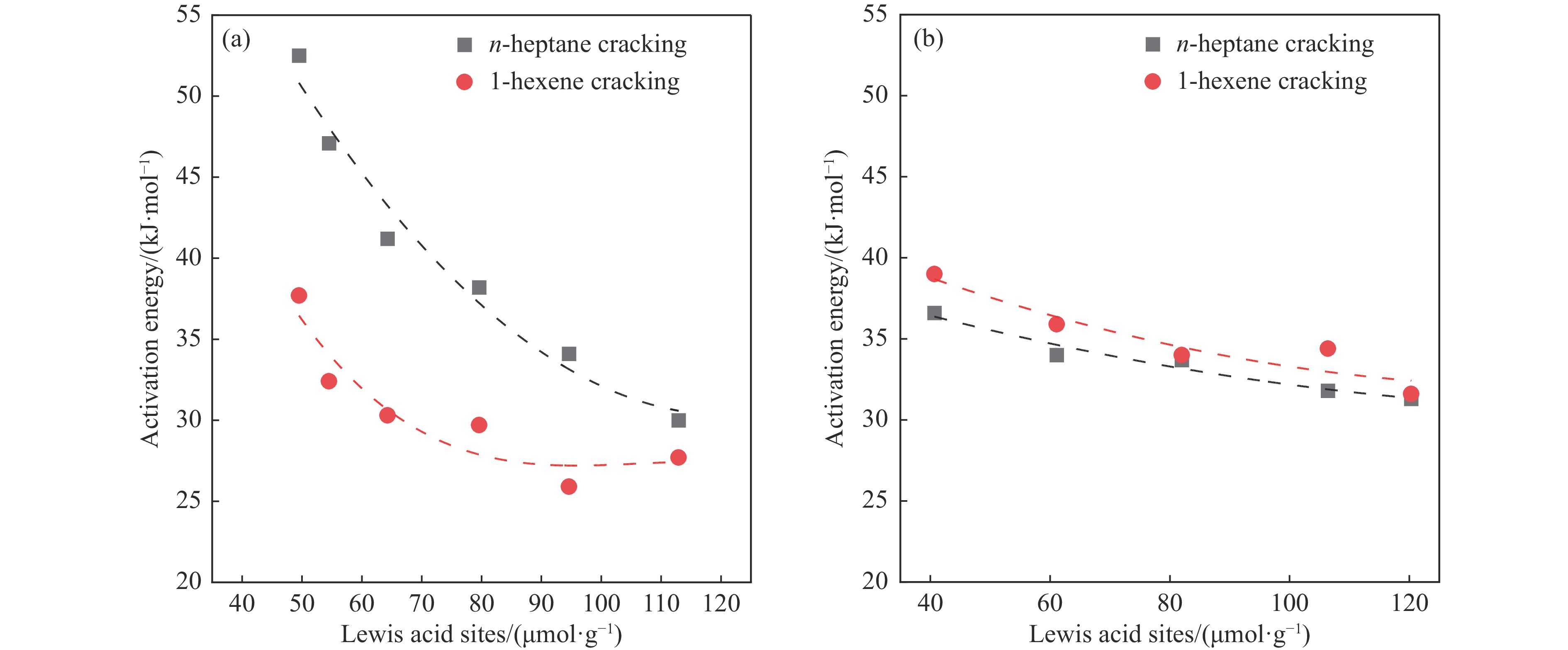
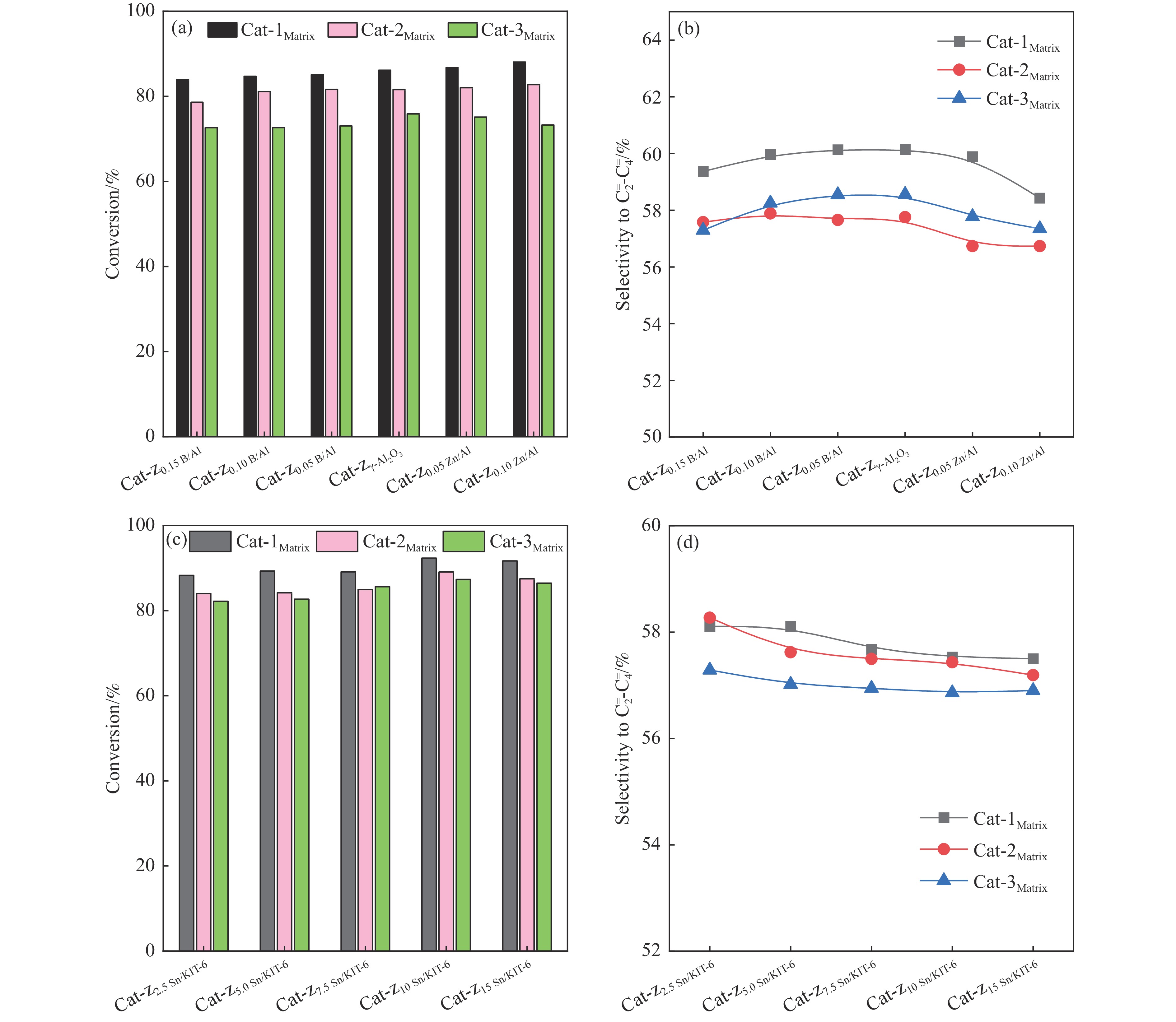
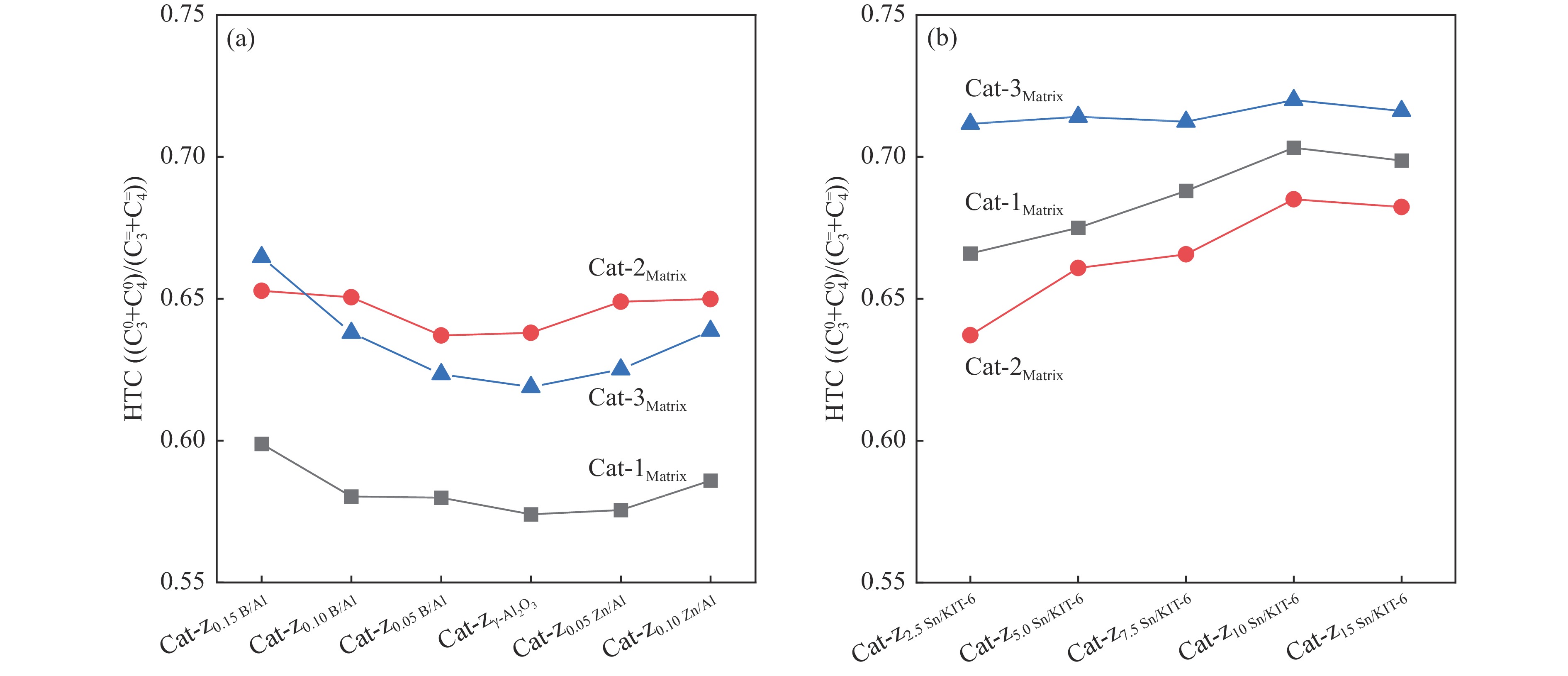
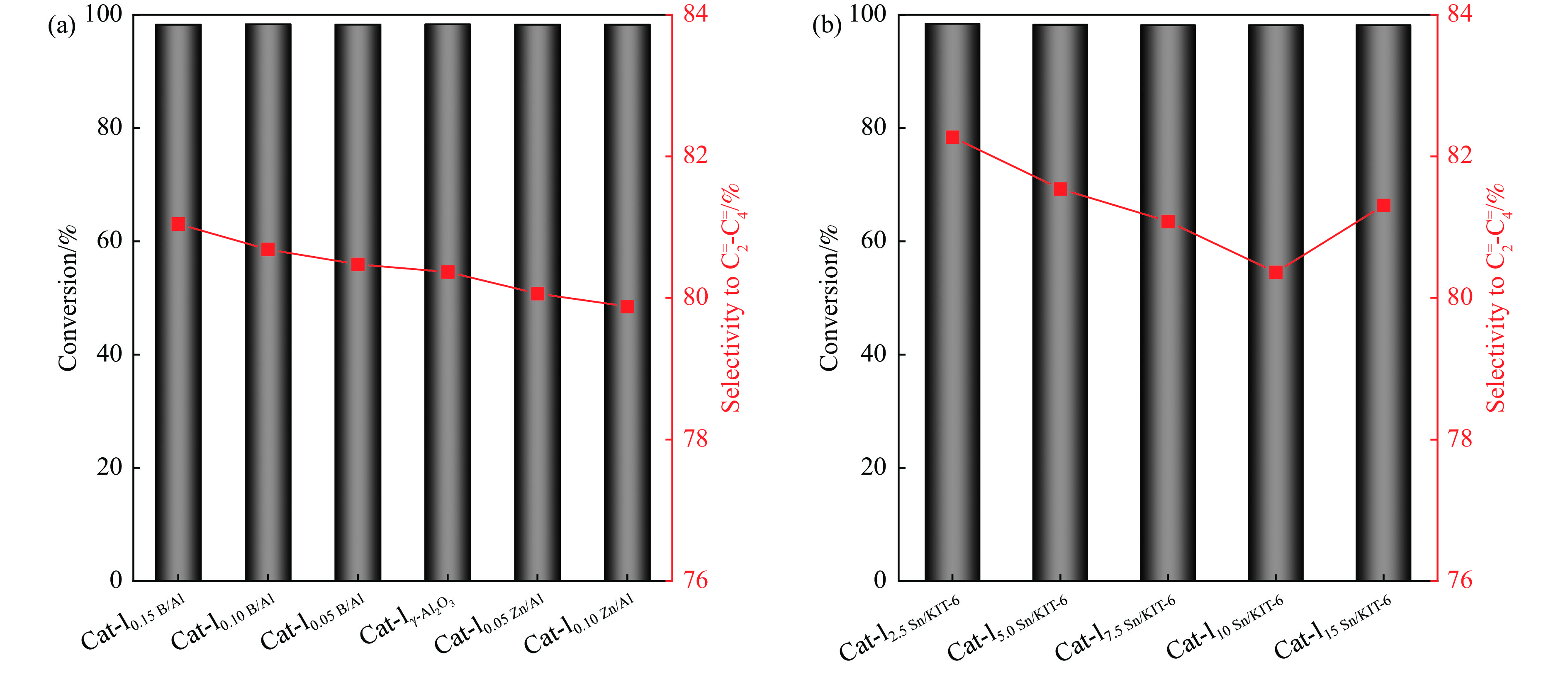
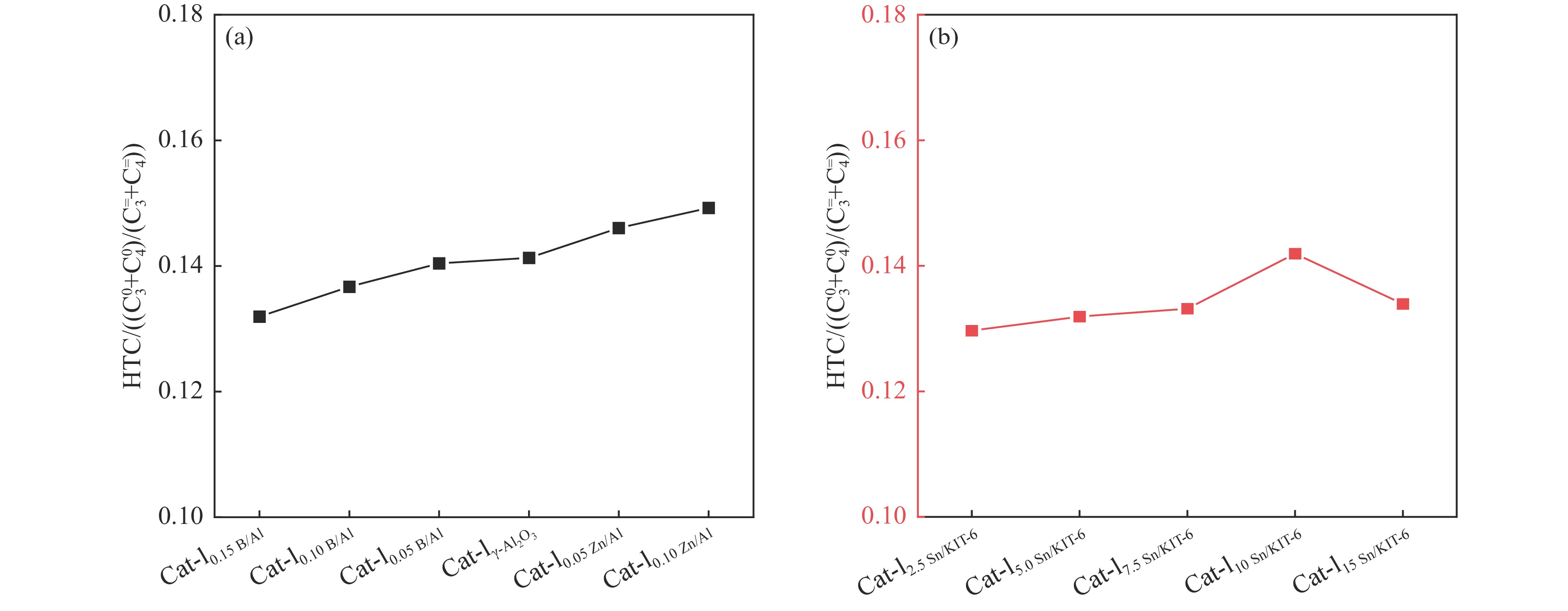
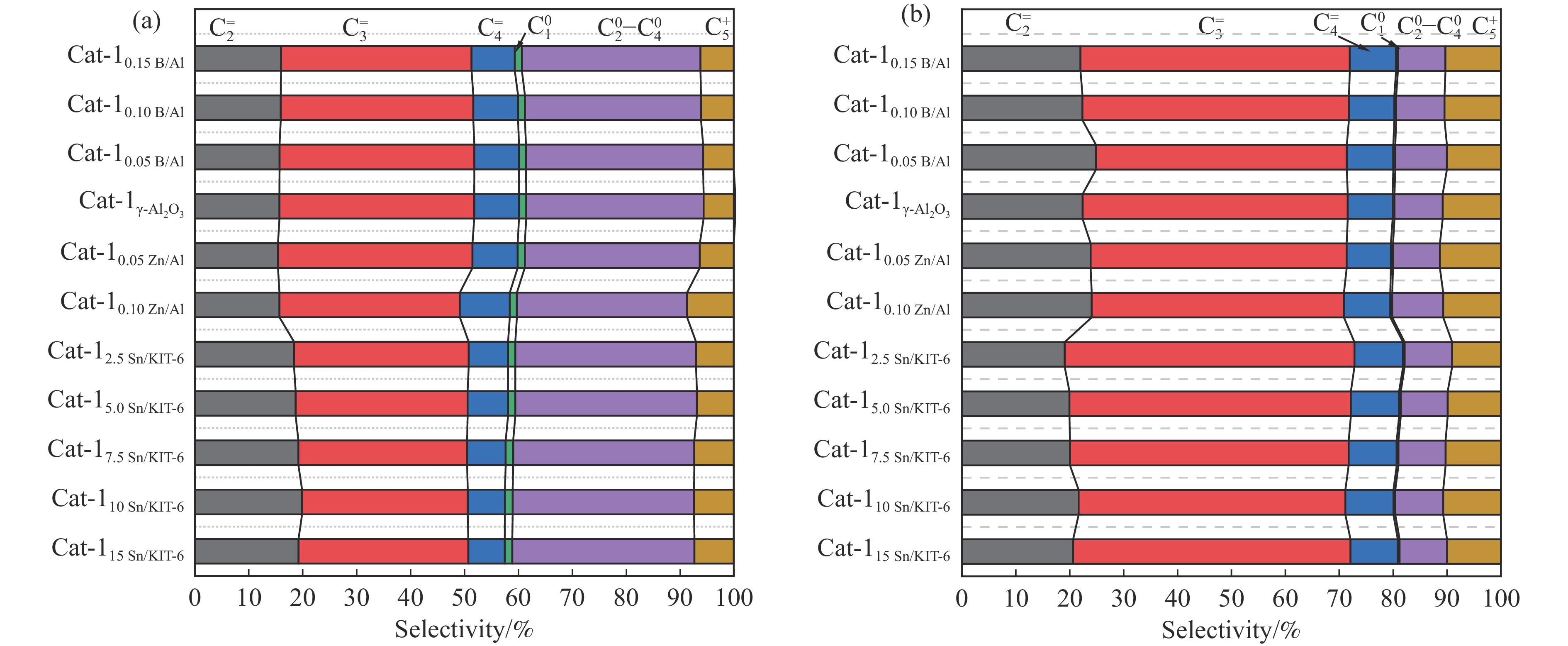
摘要: The reasonable matching of zeolite and matrix is one of the most effective strategies to increase the yield of light olefins in naphtha catalytic cracking. However, the influence of the surface Lewis acidity within the matrix on the cracking reactions has remained ambiguous. Therefore, in present study, boron and zinc co-modified γ-Al2 O3 and tin modified mesoporous silica KIT-6 with tuned surface Lewis acidity were applied to evaluate the cracking reactivity of n -heptane and 1-hexene to light olefins, in which the matrix was used alone and coupled with ZSM-5 zeolite in different packed modes. The effects of the modifiers on the textural properties and surface acidity of γ-Al2 O3 and KIT-6 were investigated by XRD, TEM, N2 physical absorption-desorption, and NH3 -TPD. The results showed that B doping reduced the Lewis acidity (both in the amount and acid strength) of γ-Al2 O3 , while the incorporation of Zn doping led to increased Lewis acidity. In addition, the Lewis acidity of ordered mesoporous KIT-6 increased as Sn doping rose. While for pure matrix, the ascend in conversions of n -heptane and 1-hexene was consistent with the increased Lewis acidity of the B and Zn co-modified γ-Al2 O3 and x Sn/KIT-6 rose, along with decreased activation energy. In contrast, when coupled with ZSM-5 zeolite, the highest conversion was achieved in the dual-bed manner of matrix and zeolite, and the conversion increased concomitantly with the increase in the Lewis acidity of the matrix. However, excessive Lewis acidity can accelerate the hydrogen transfer rate while diminishing the selectivity of light olefins.
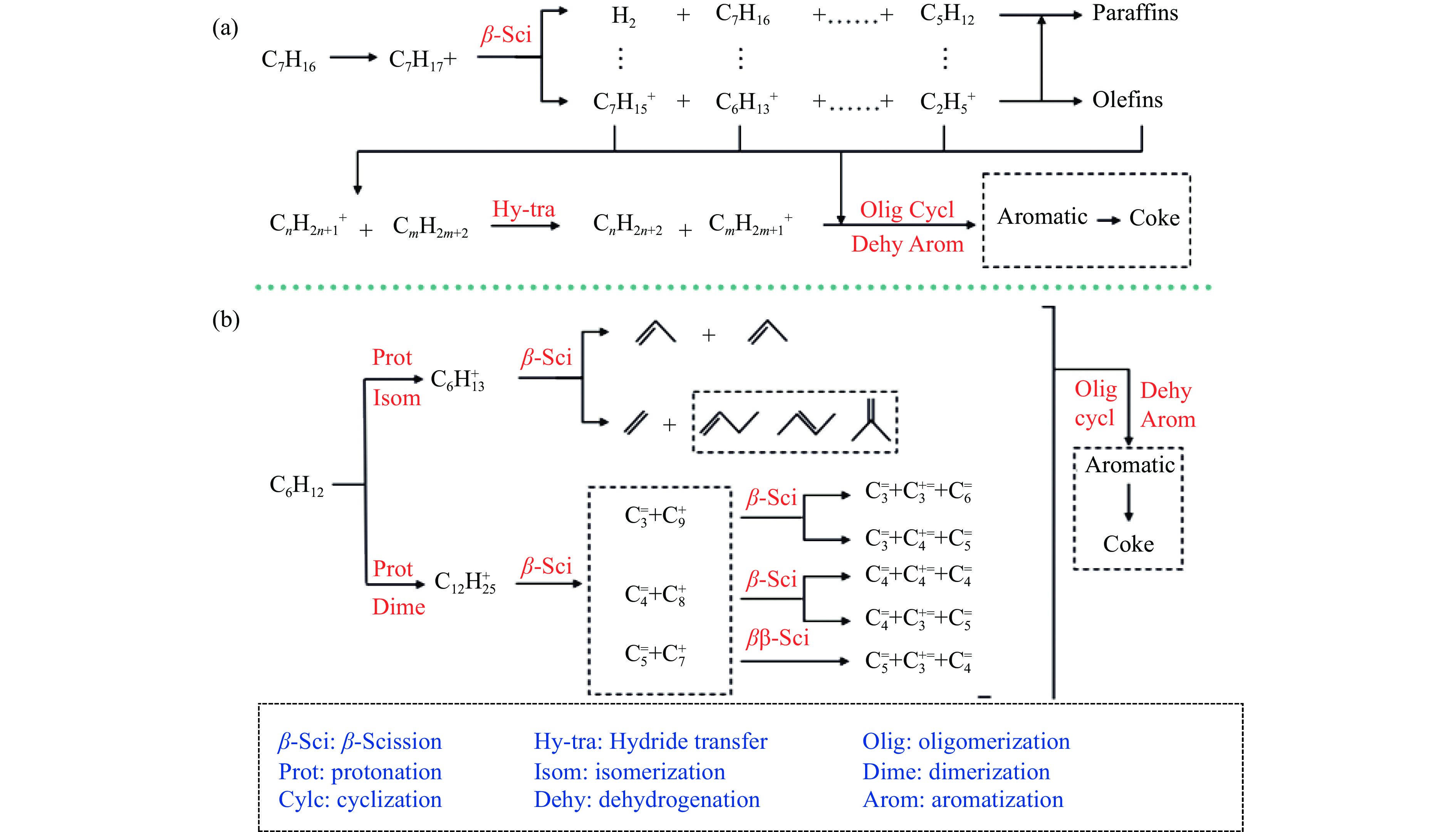
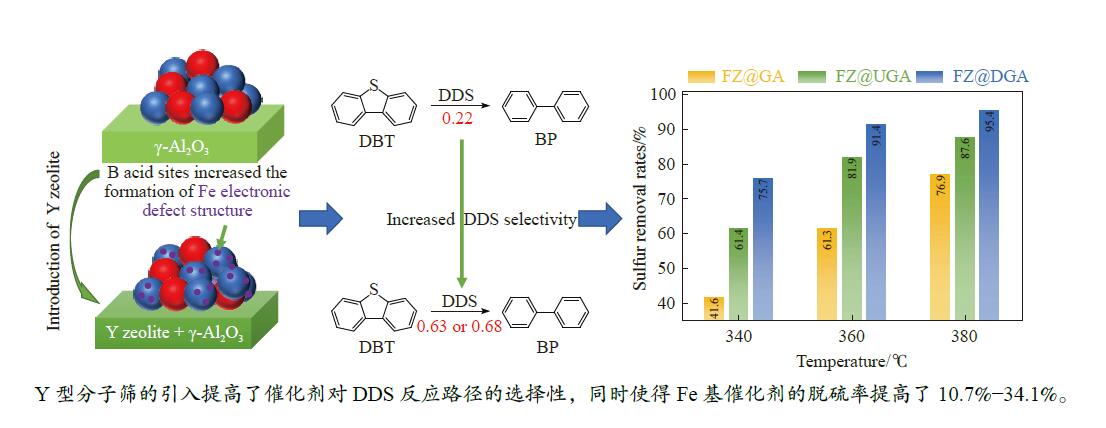
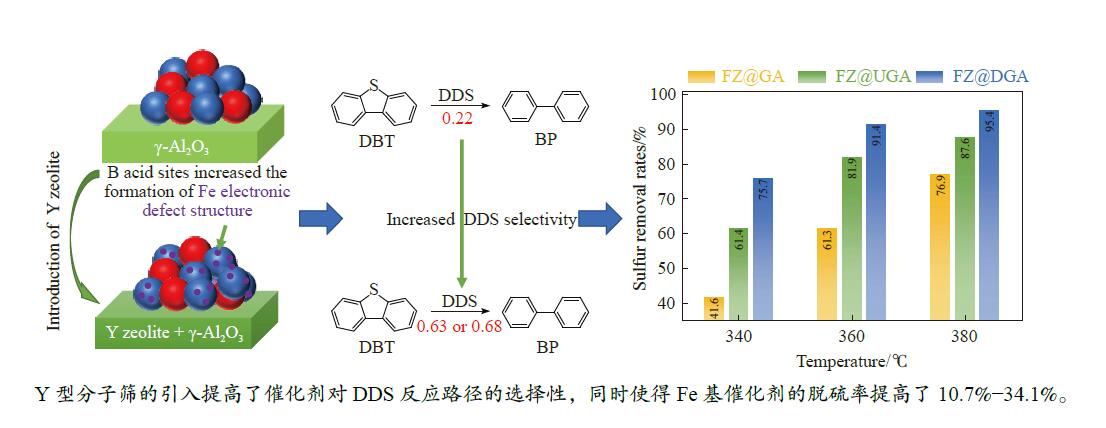
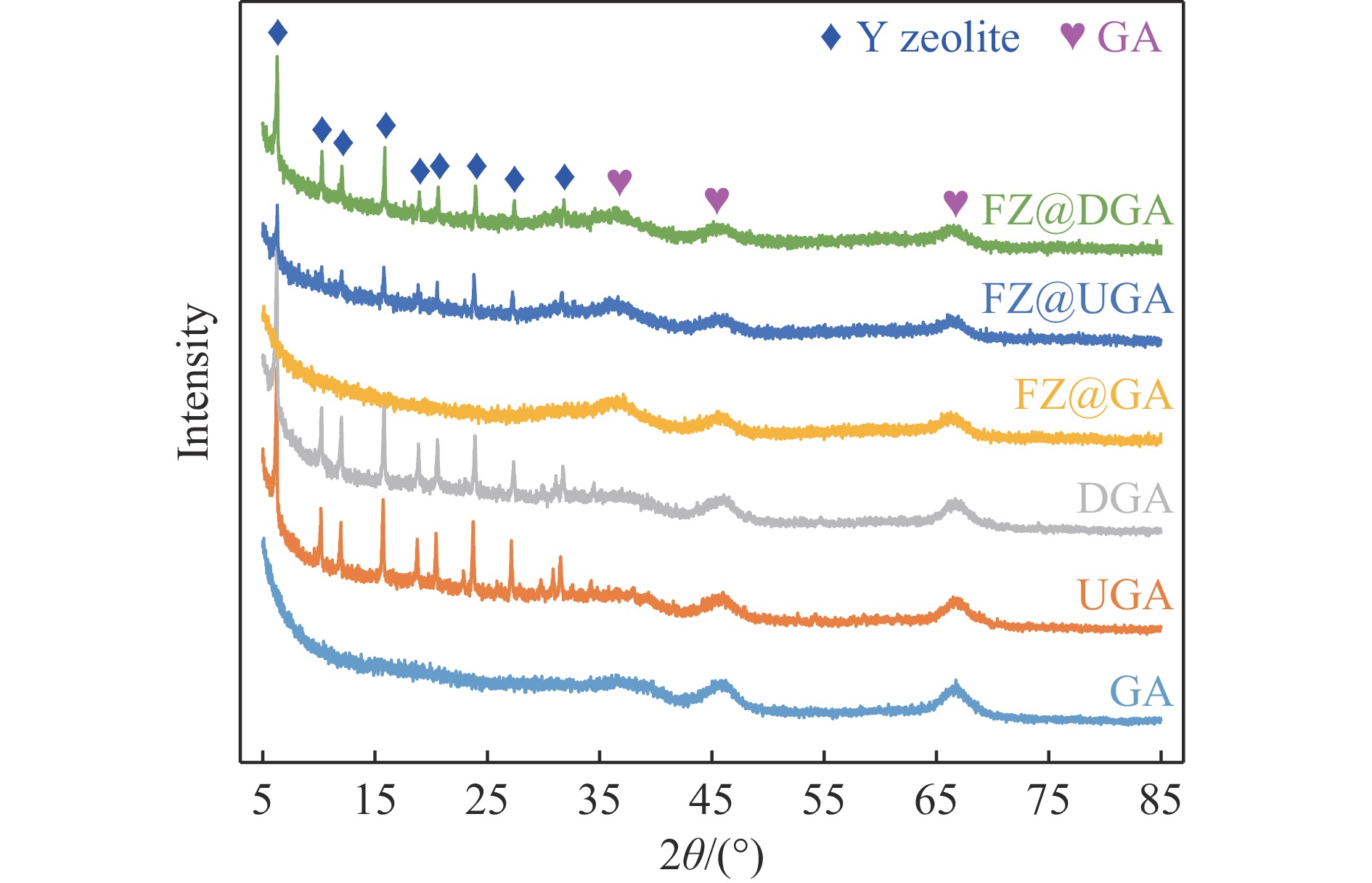
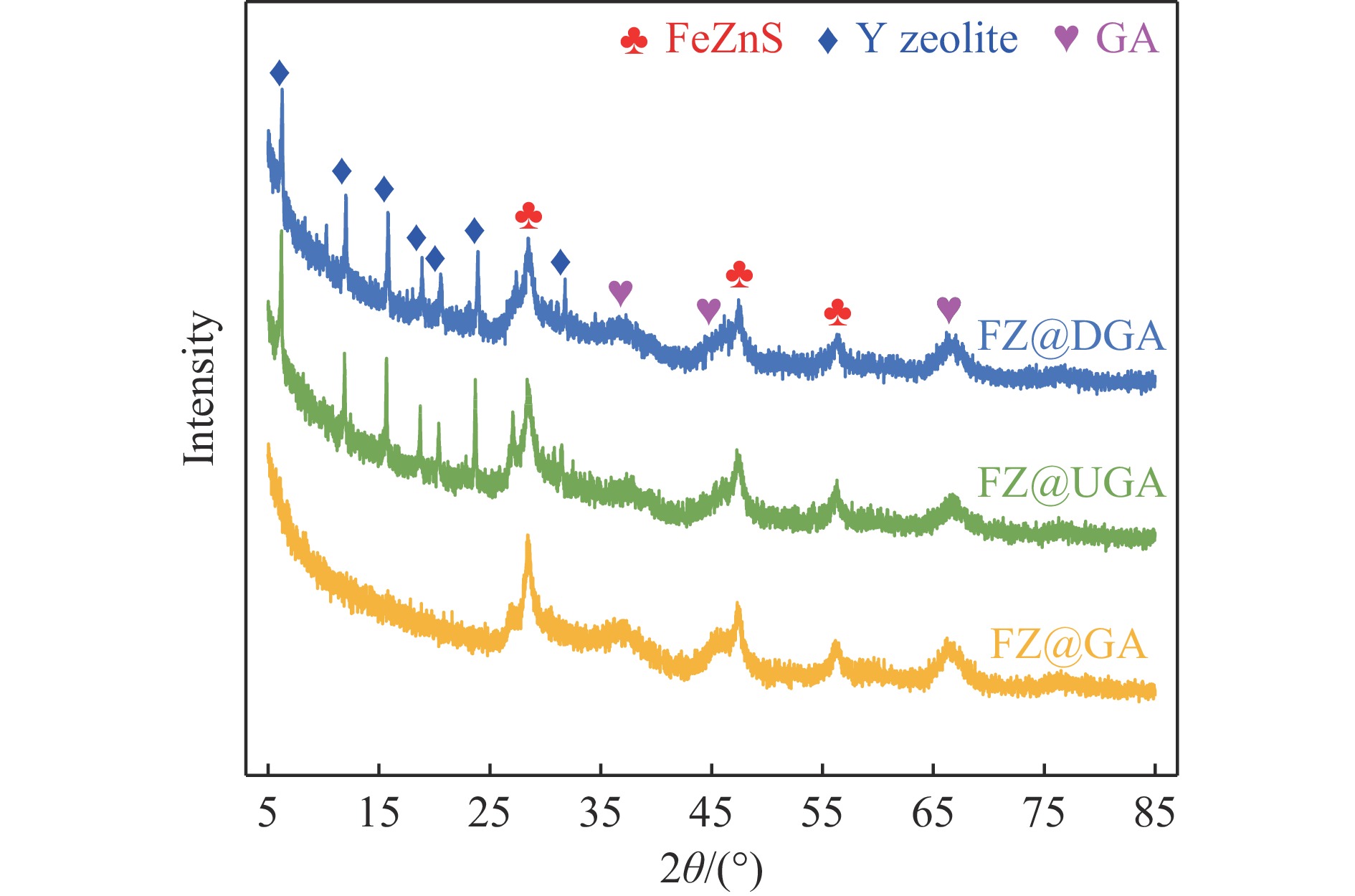
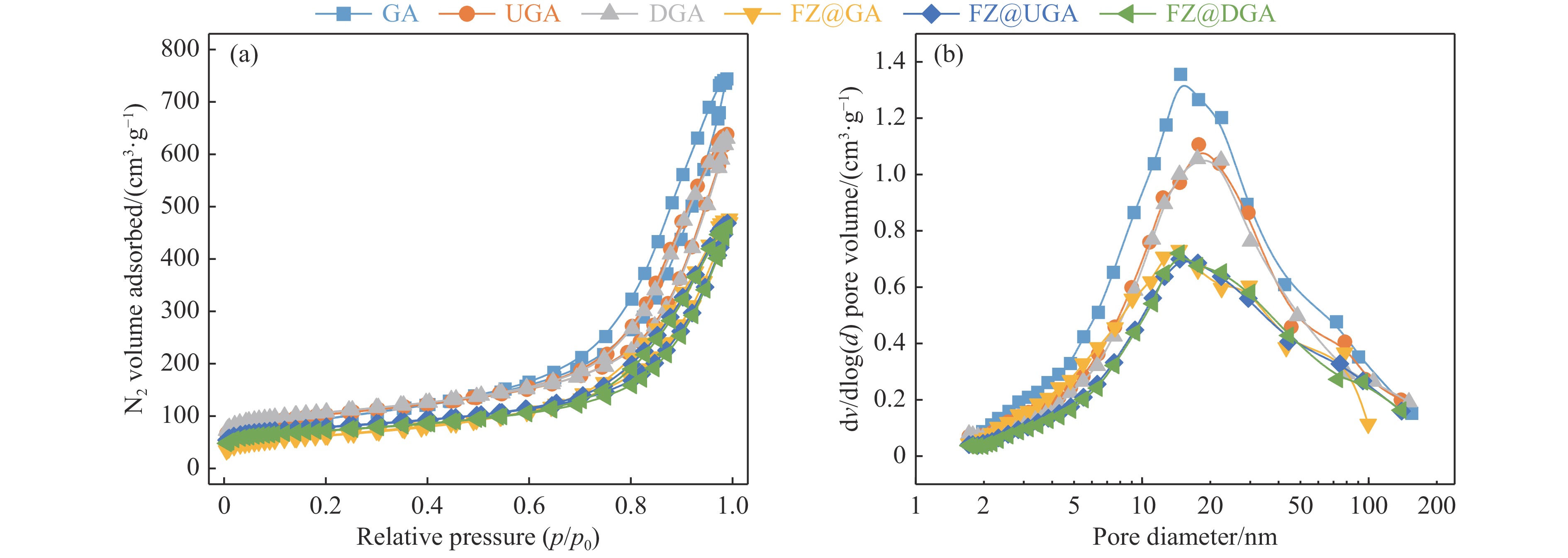
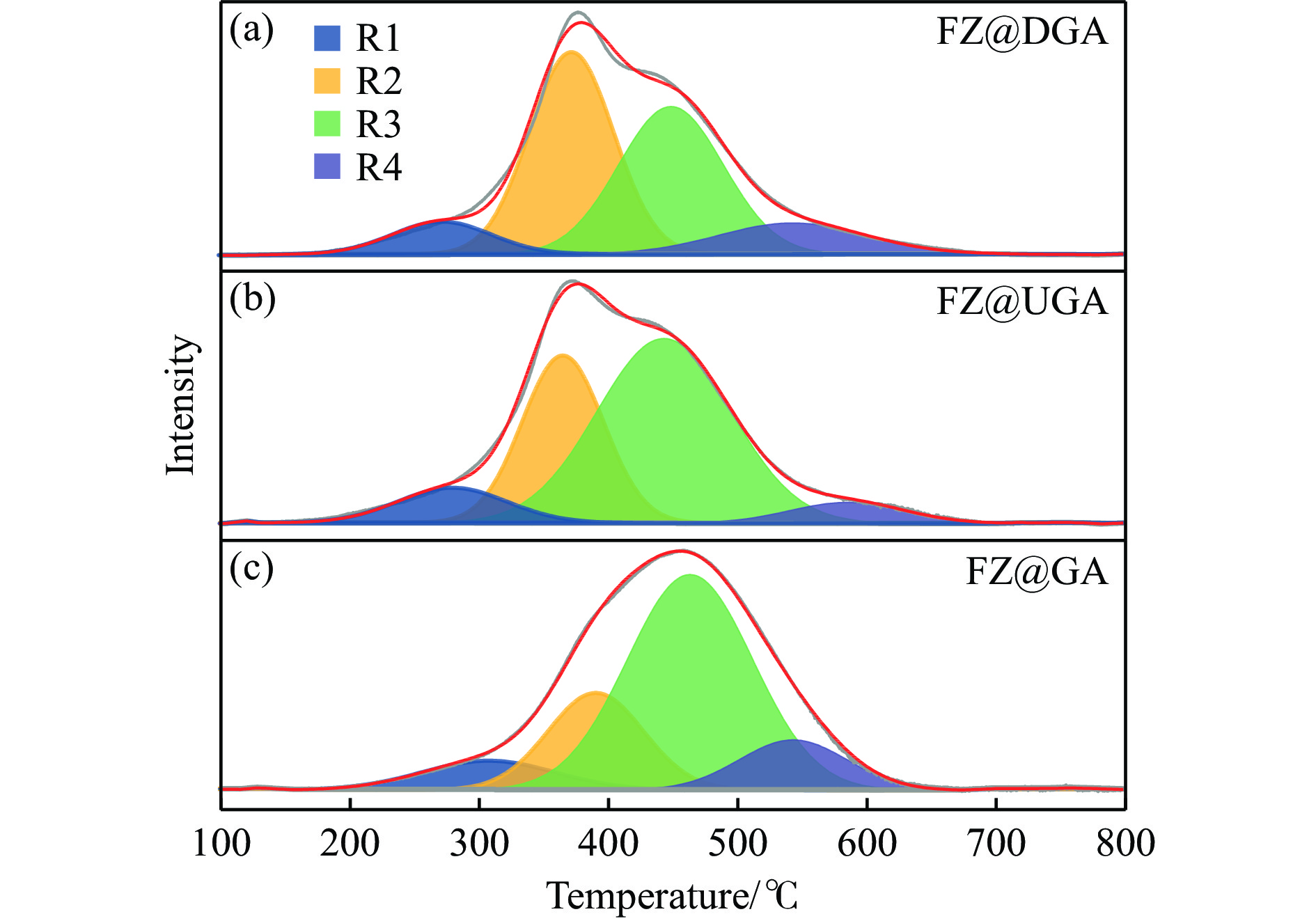
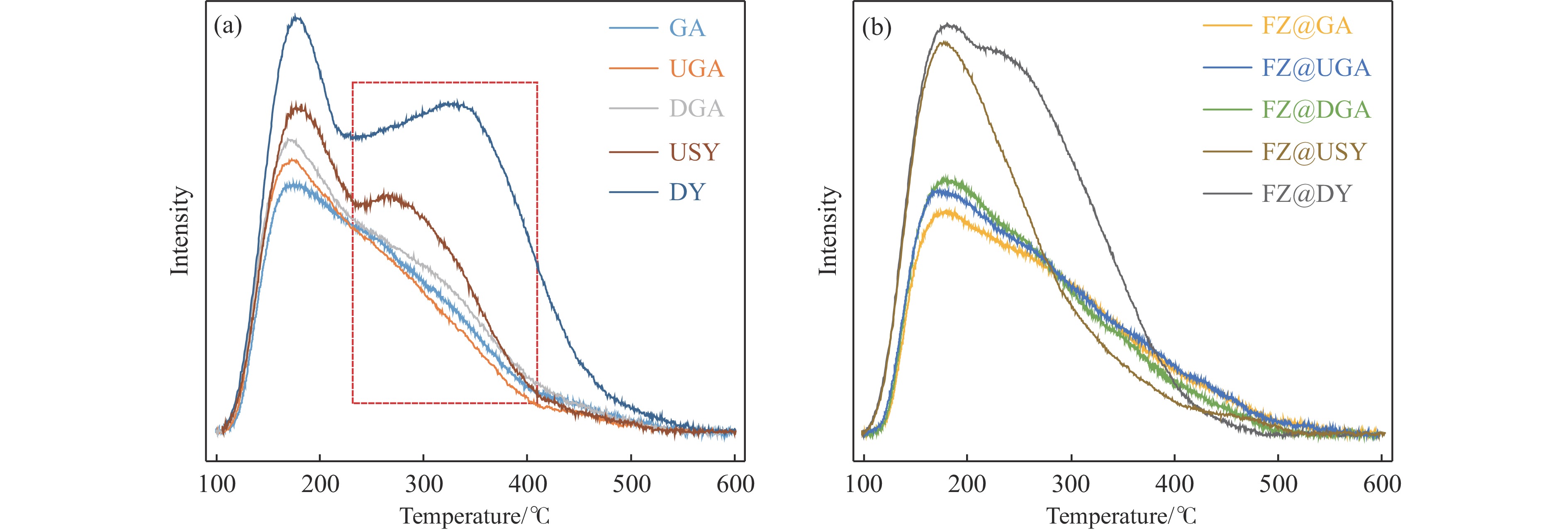
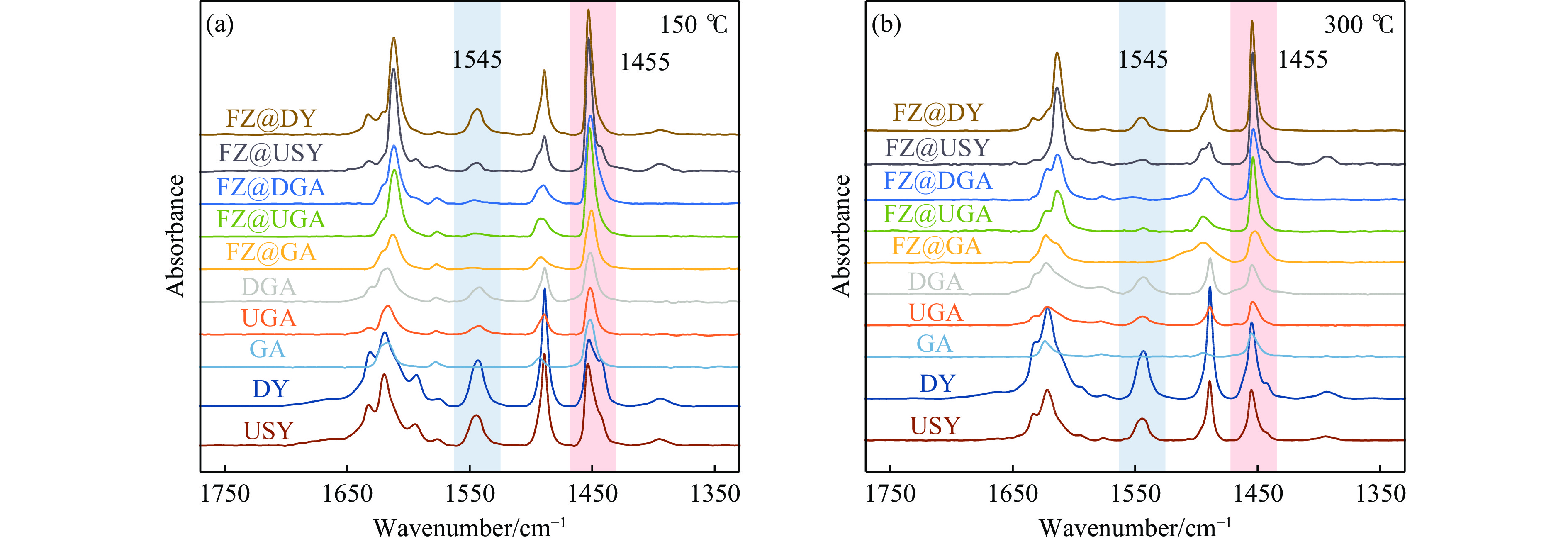
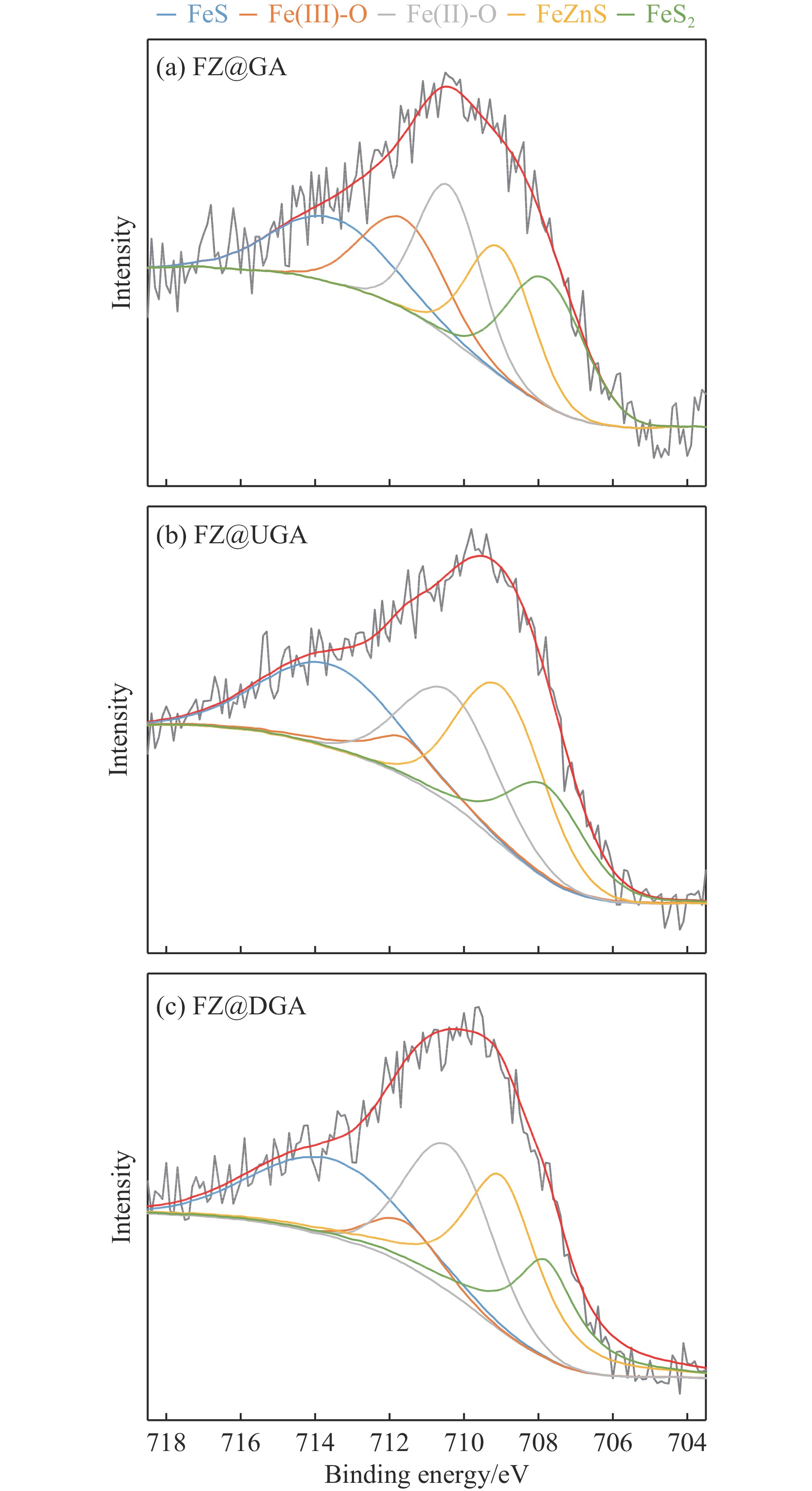
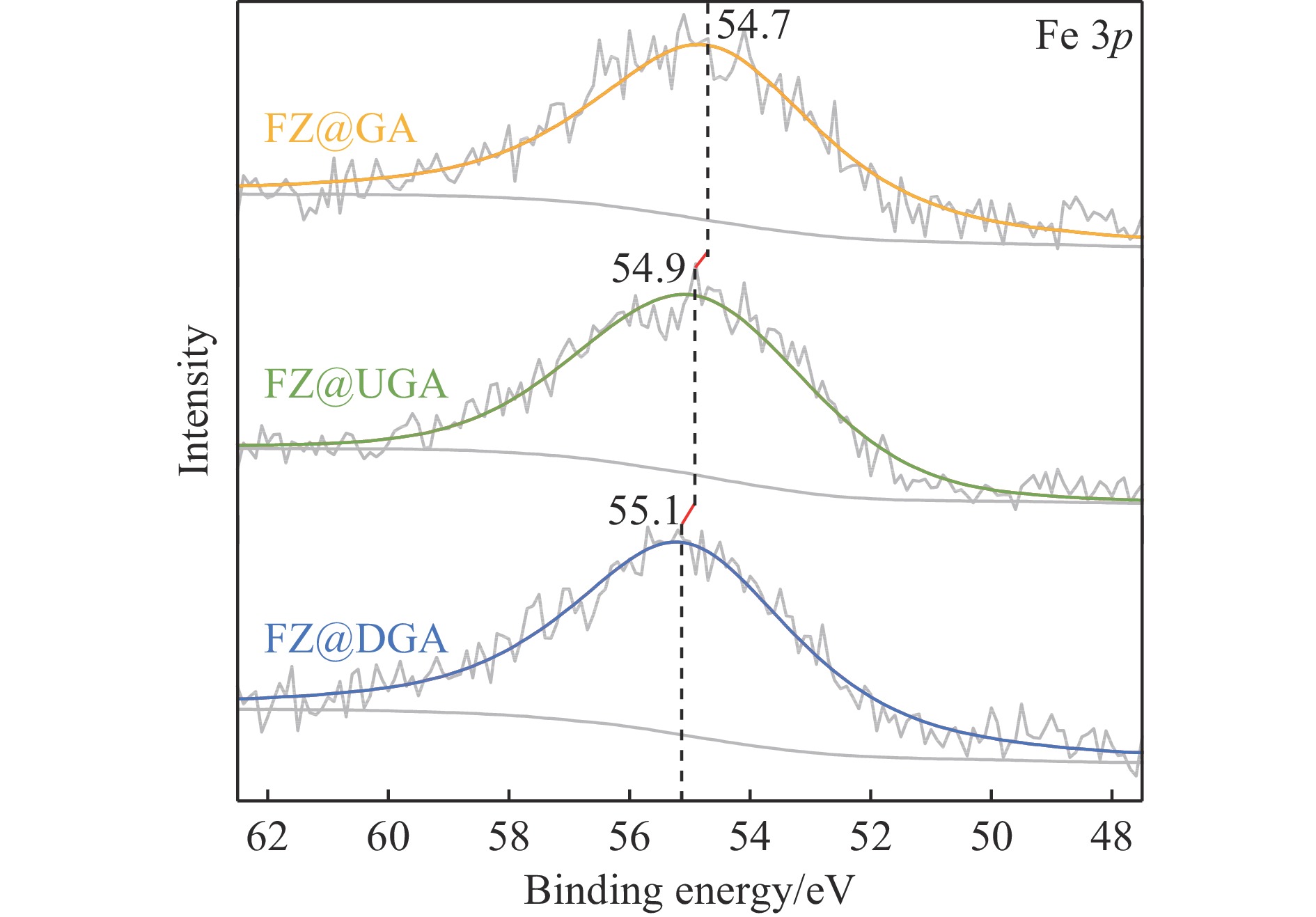
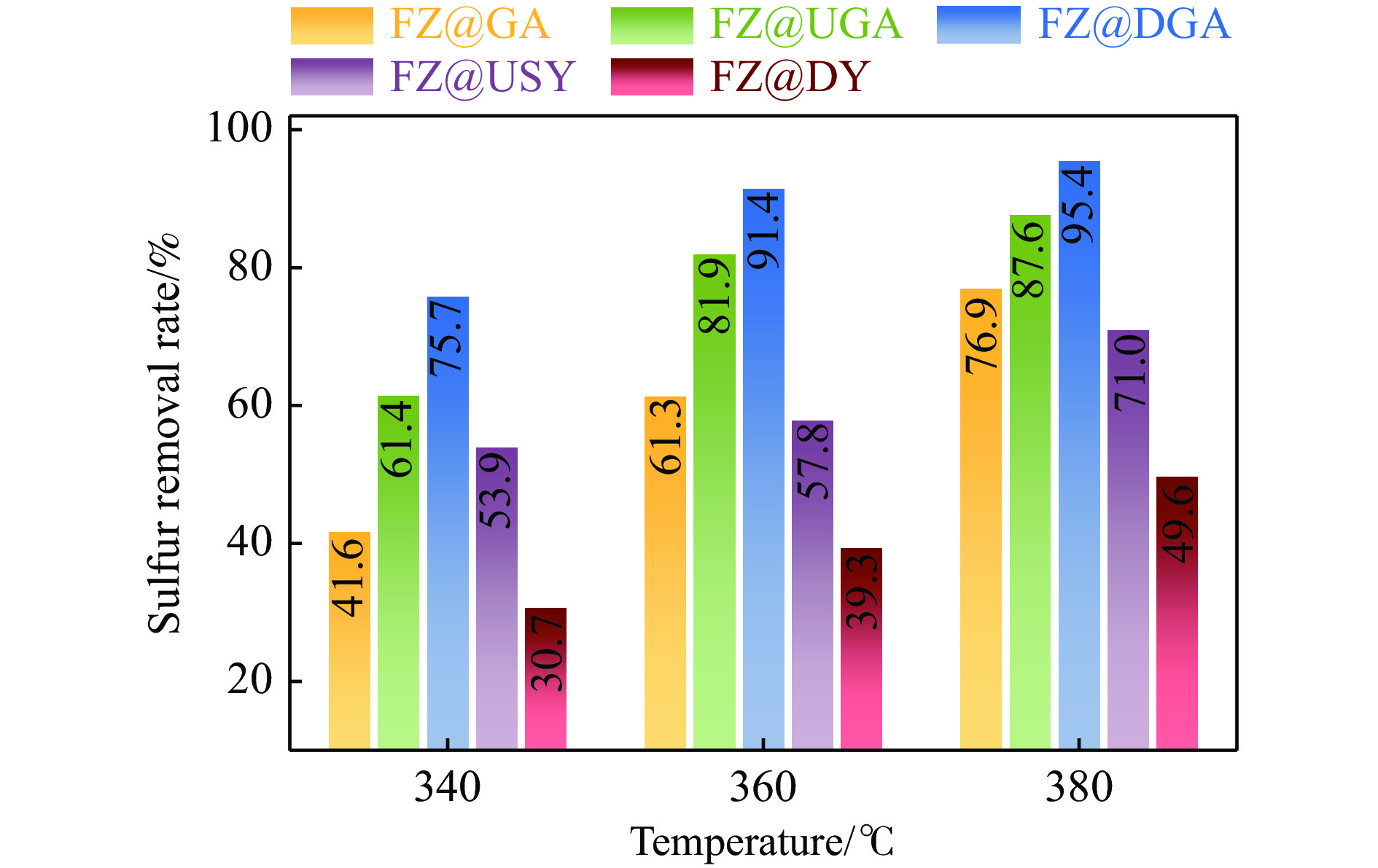
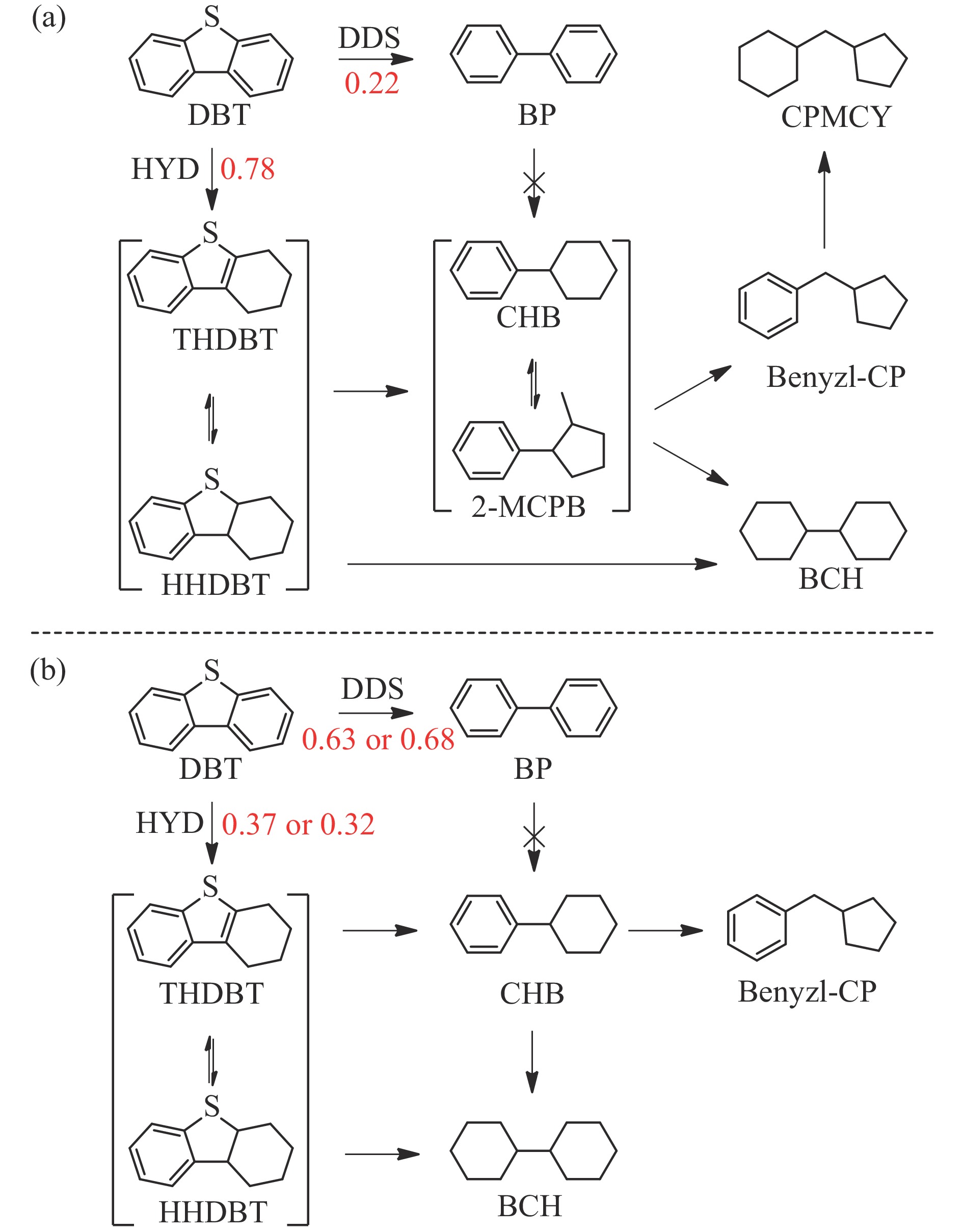
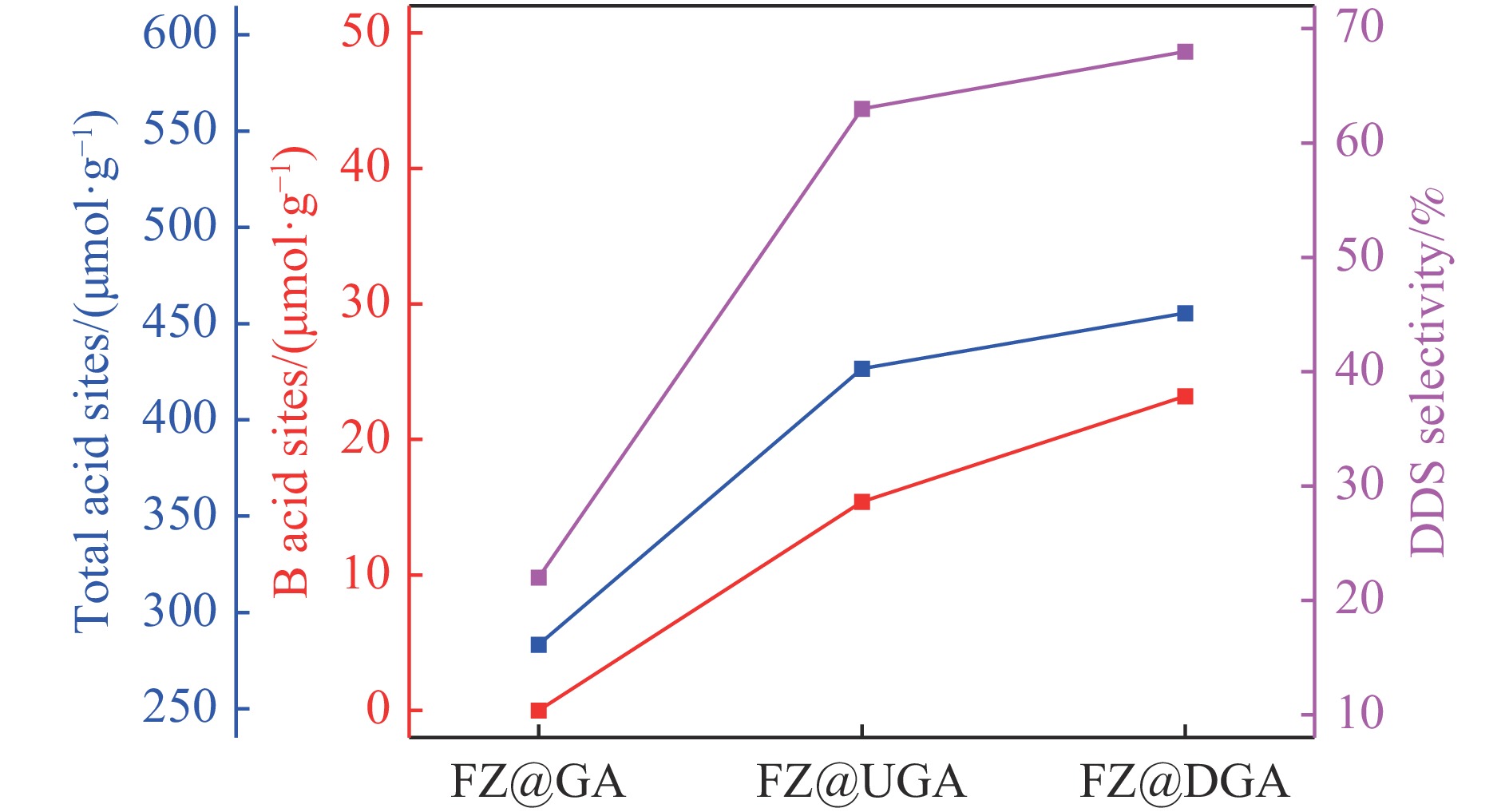
摘要: 以Fe作为主活性金属、Zn作为助活性金属,制备了Y型分子筛改性的Fe基加氢脱硫(HDS)催化剂。采用低温氮气物理吸附、X射线衍射(XRD)、氢气程序升温还原(H2 -TPR)、氨气程序升温脱附(NH3 -TPD)、扫描电子显微镜(SEM)、X射线光电子能谱(XPS)和吡啶红外(Py-IR)等表征方法对改性前后Fe基催化剂的形貌、孔结构、分散性、还原性、电子缺陷结构以及酸性等变化进行了研究,并使用固定床反应器对Fe基催化剂的HDS性能进行了评价。结果表明,Y型分子筛的引入提供了Brønsted(B)酸中心,使得Fe基催化剂的脱硫率提高了10.7%−34.1%。同时,B酸中心提高了催化剂的直接脱硫(DDS)反应路径的选择性。此外,B酸中心在促进DDS反应路径选择性增加的同时,抑制了预加氢脱硫(HYD)反应路径中四氢二苯并噻吩(THDBT)和六氢二苯并噻吩(HHDBT)更进一步的深度加氢,从而在保证脱硫率提升的同时又降低了氢耗。其根本原因可能是Y型分子筛的引入增强了催化剂的酸性,特别是B酸中心和活性金属之间的相互作用促进了电子转移,从而调节了Fe物种的电子缺陷结构,进而提升了催化剂的HDS性能。
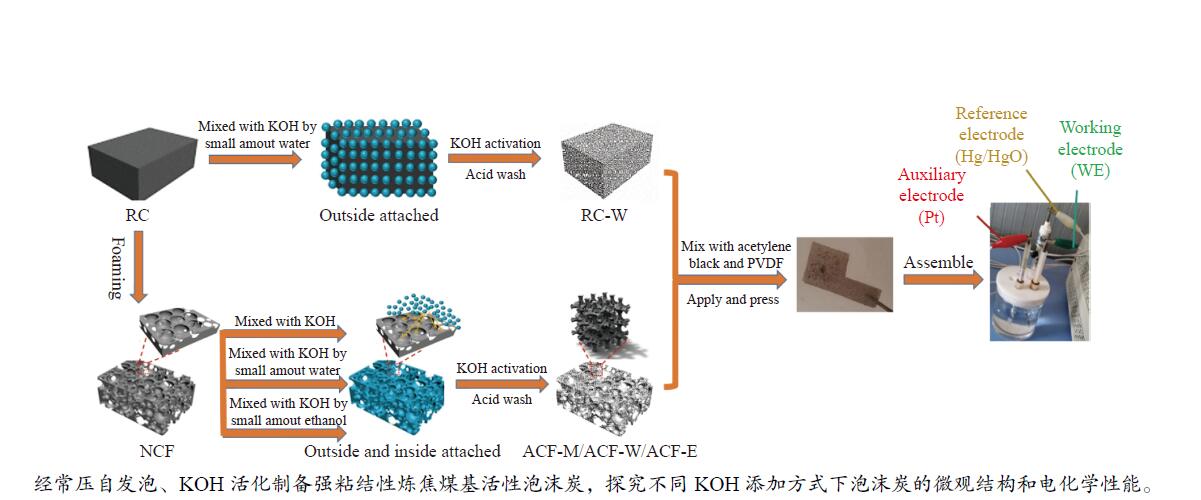
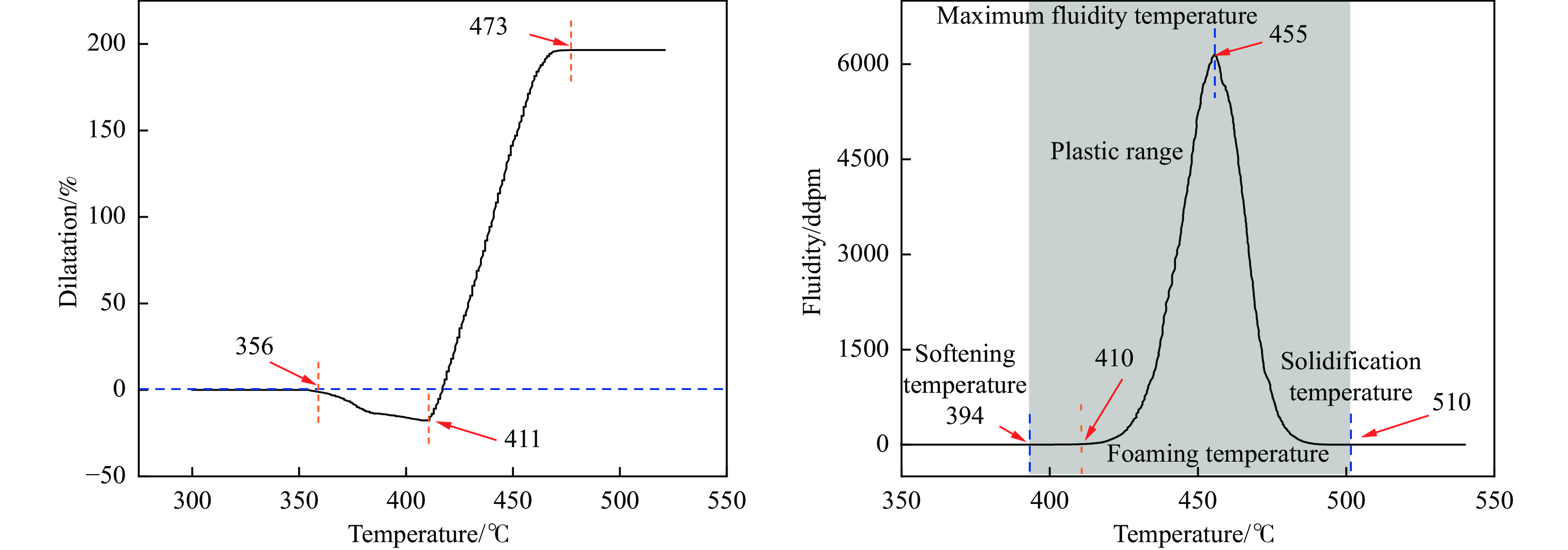
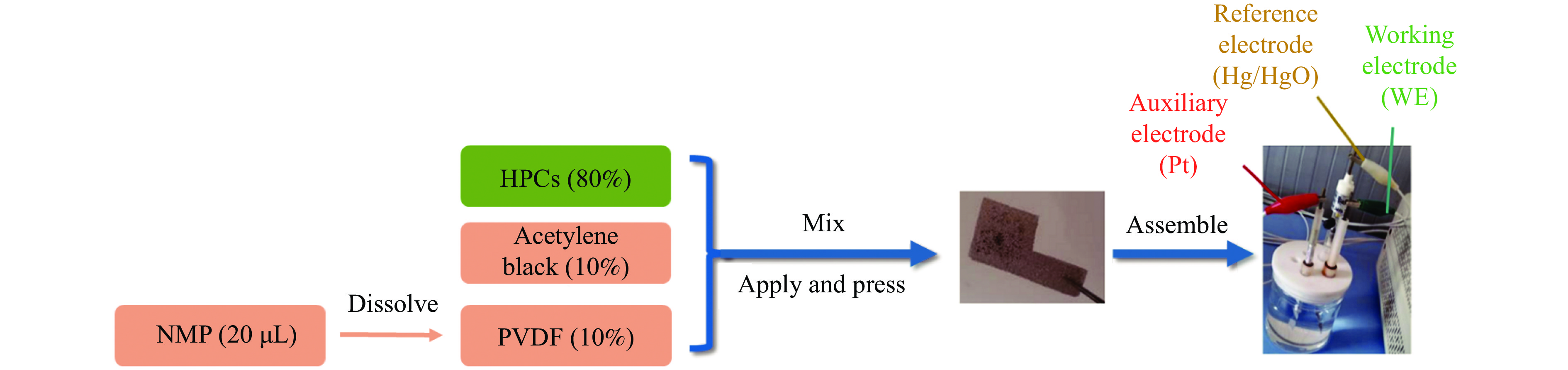
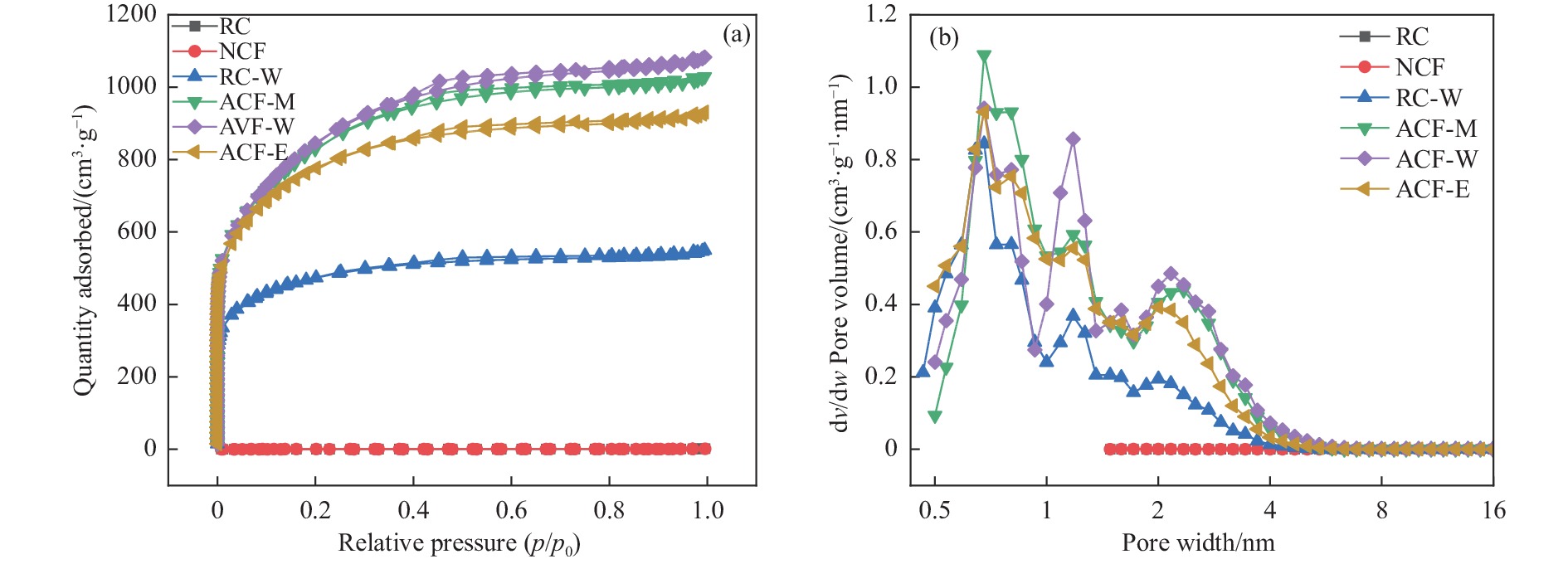
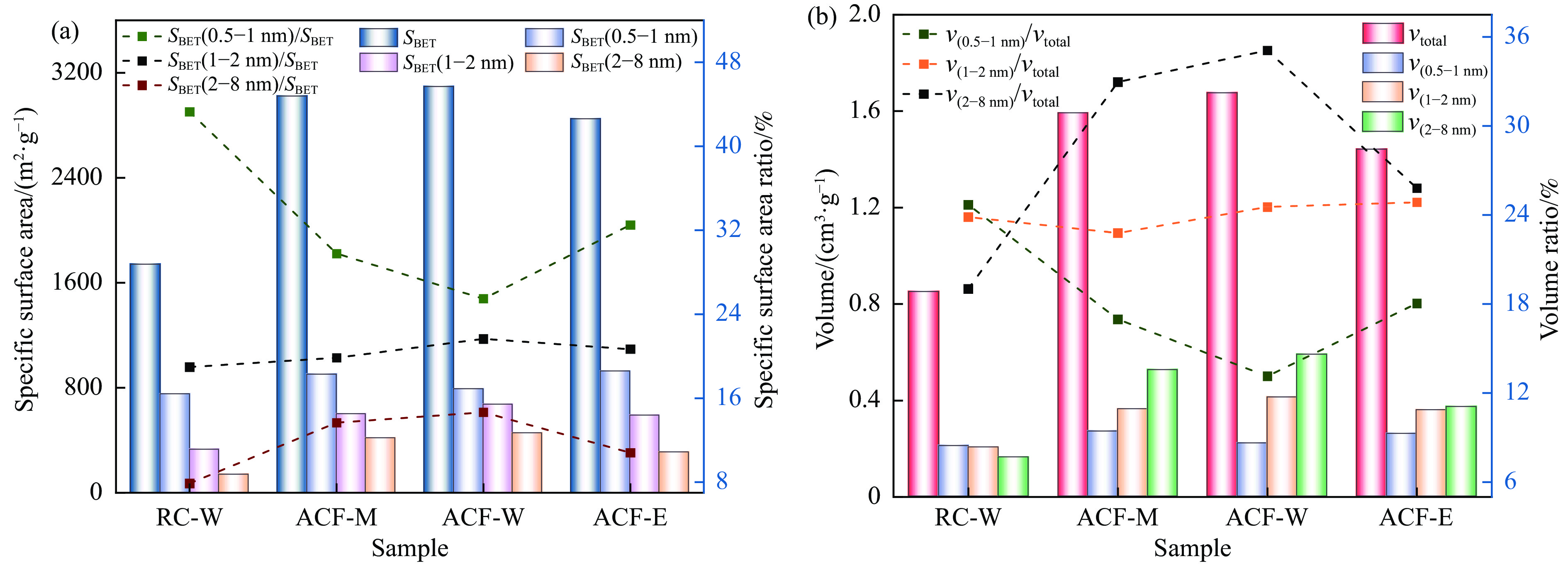
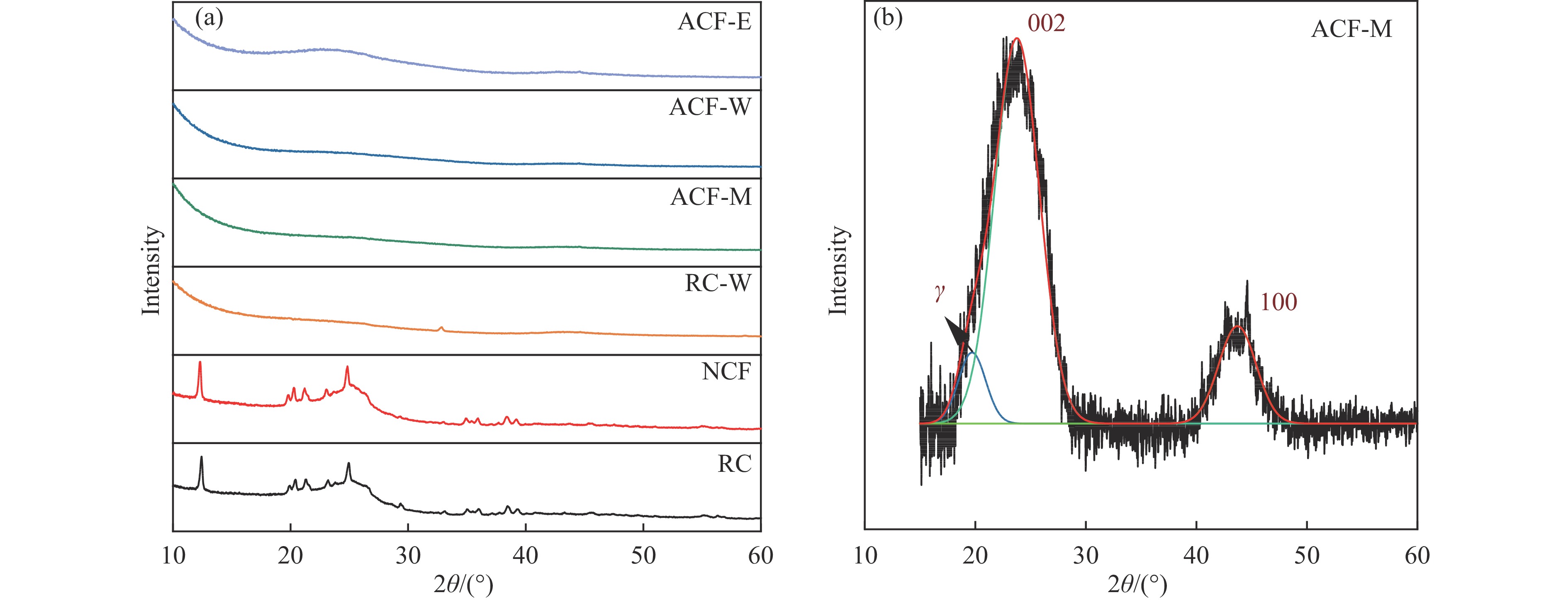
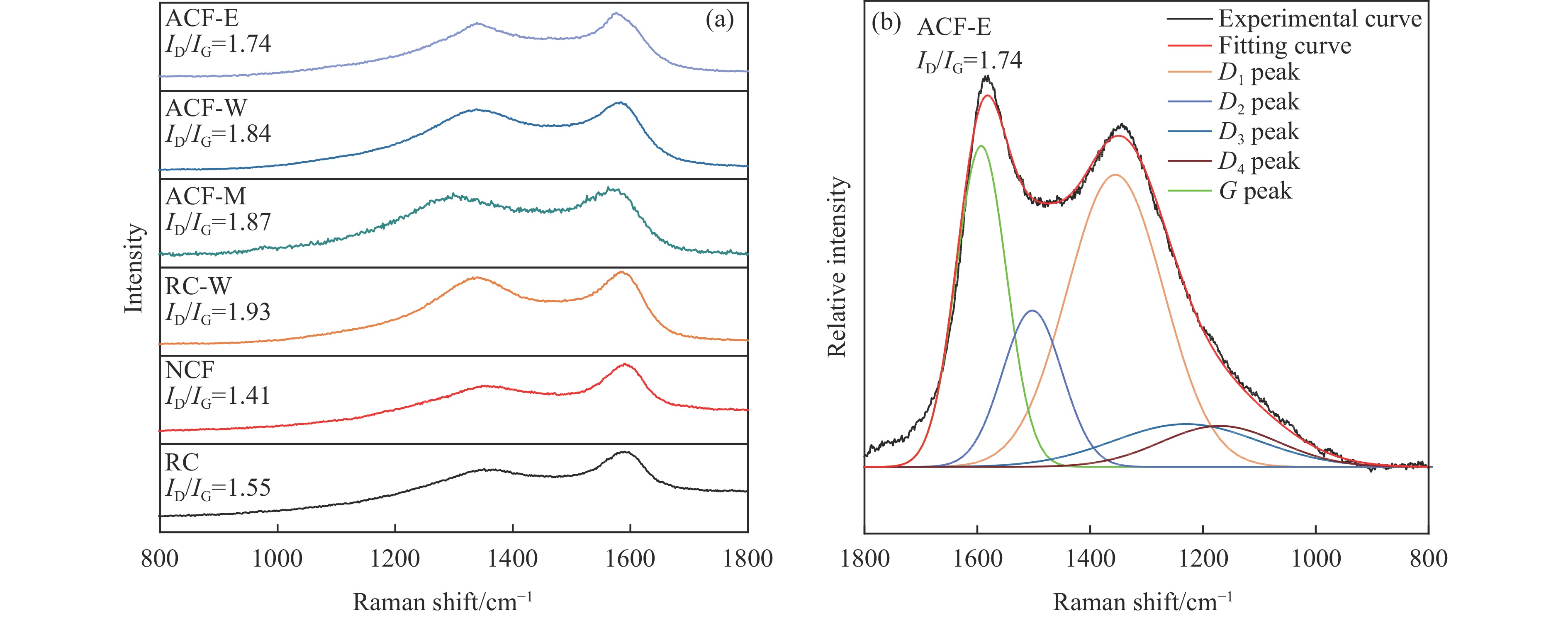
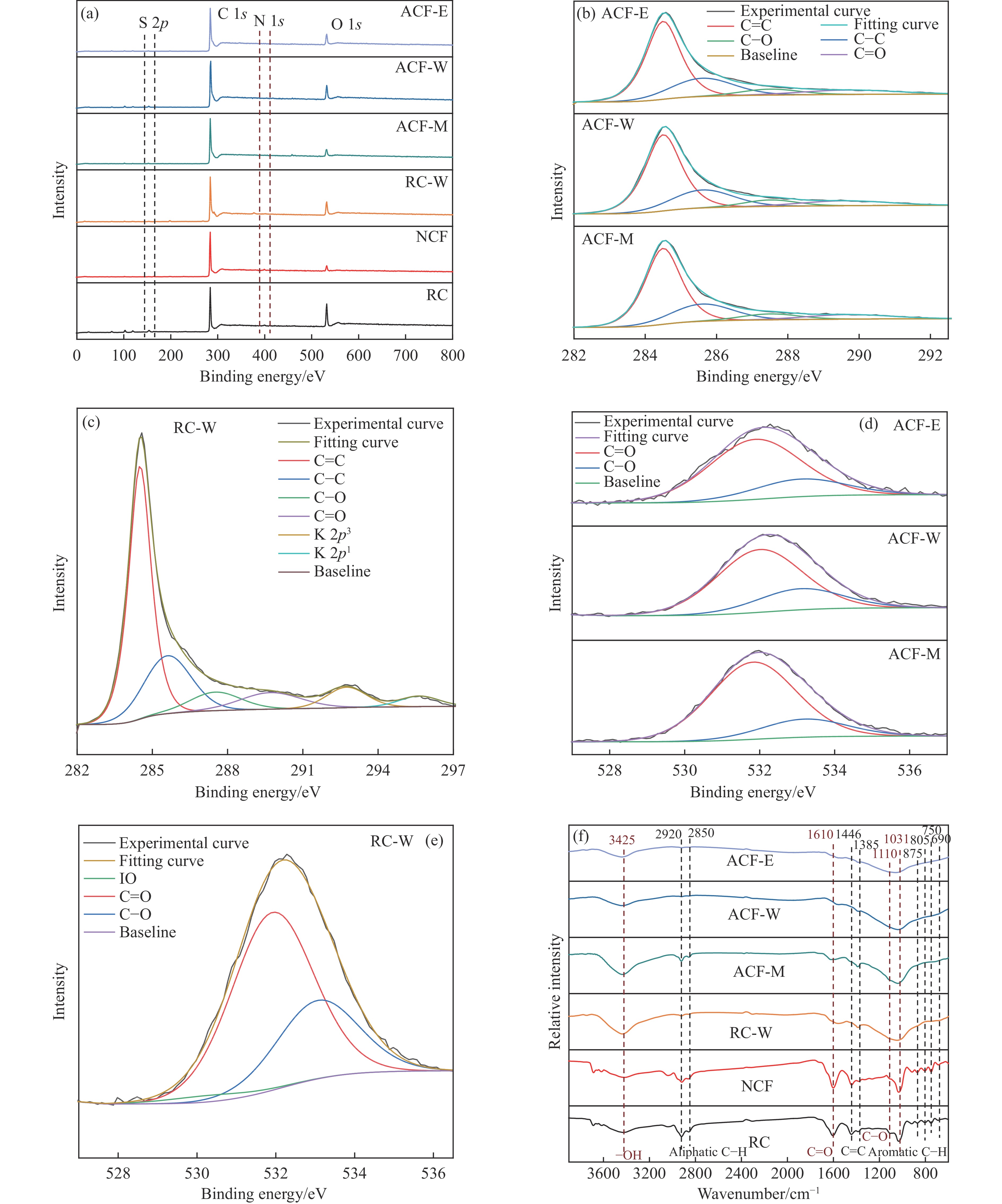
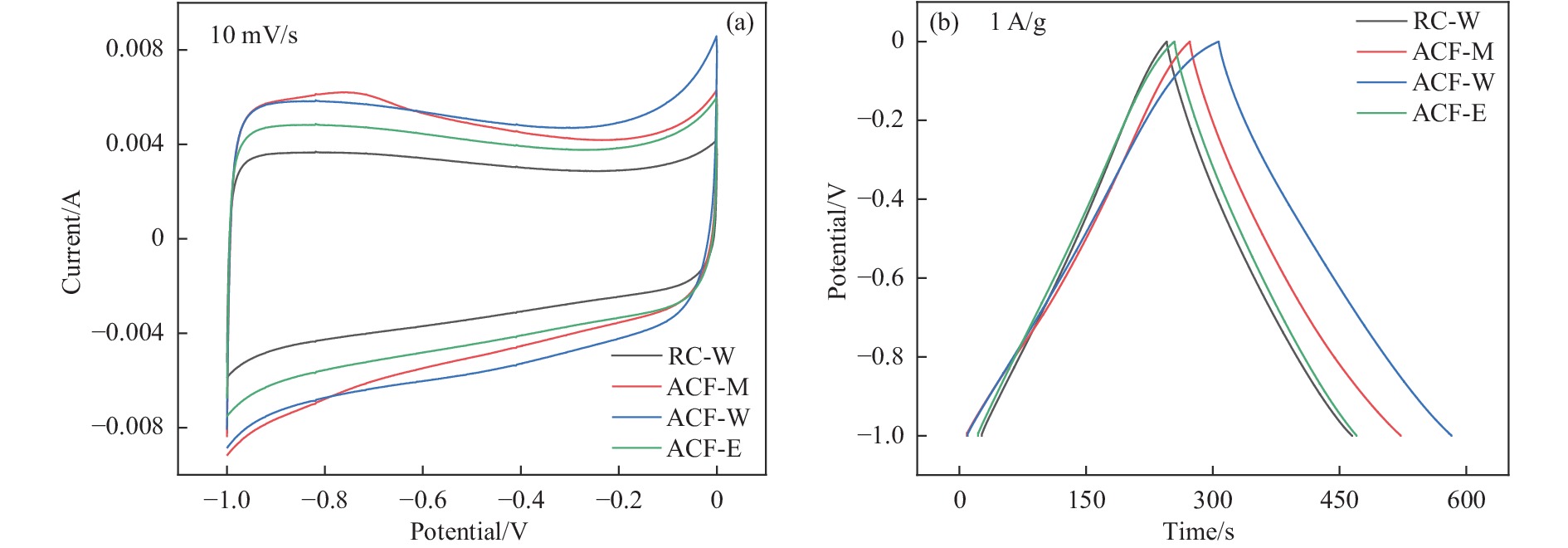
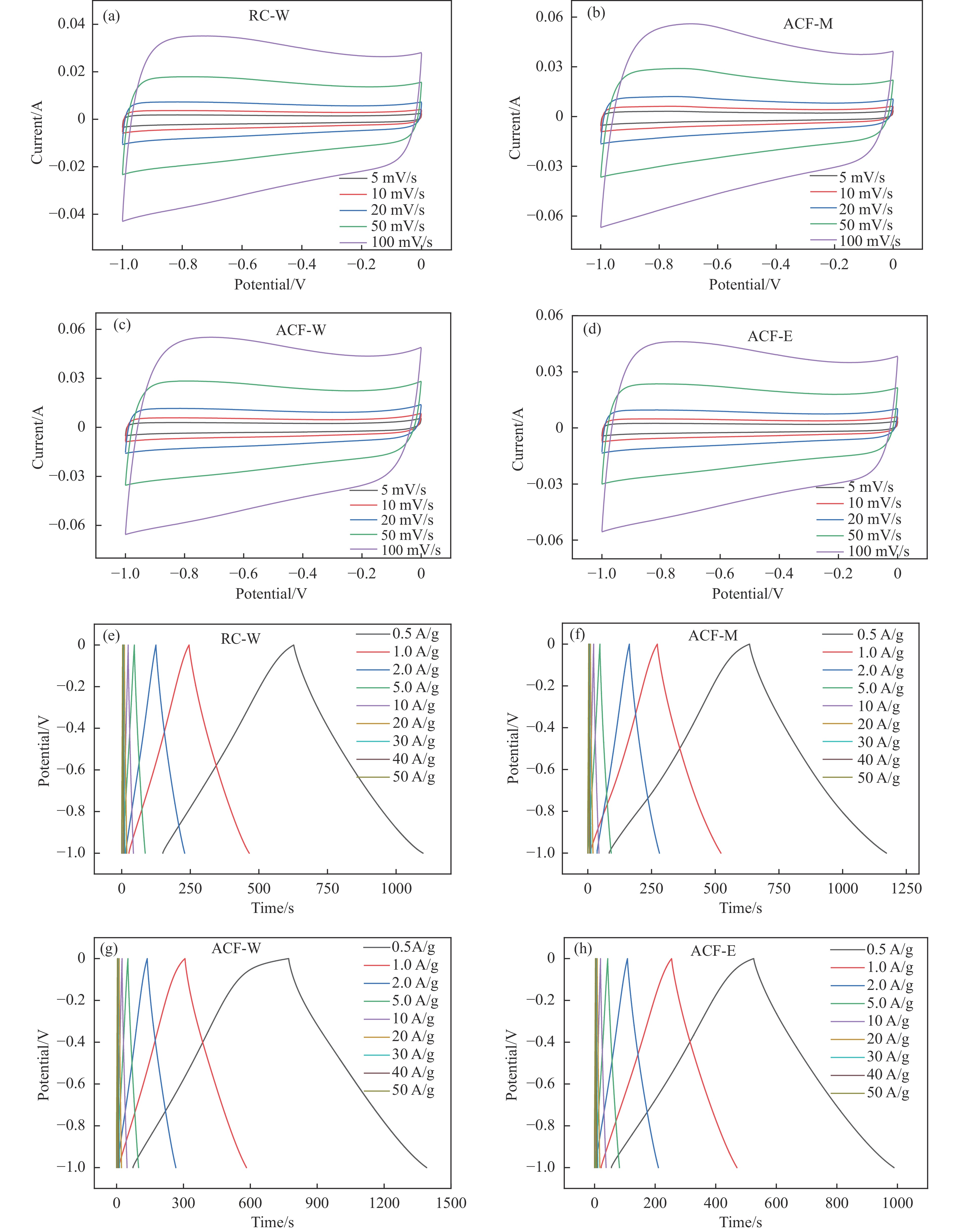
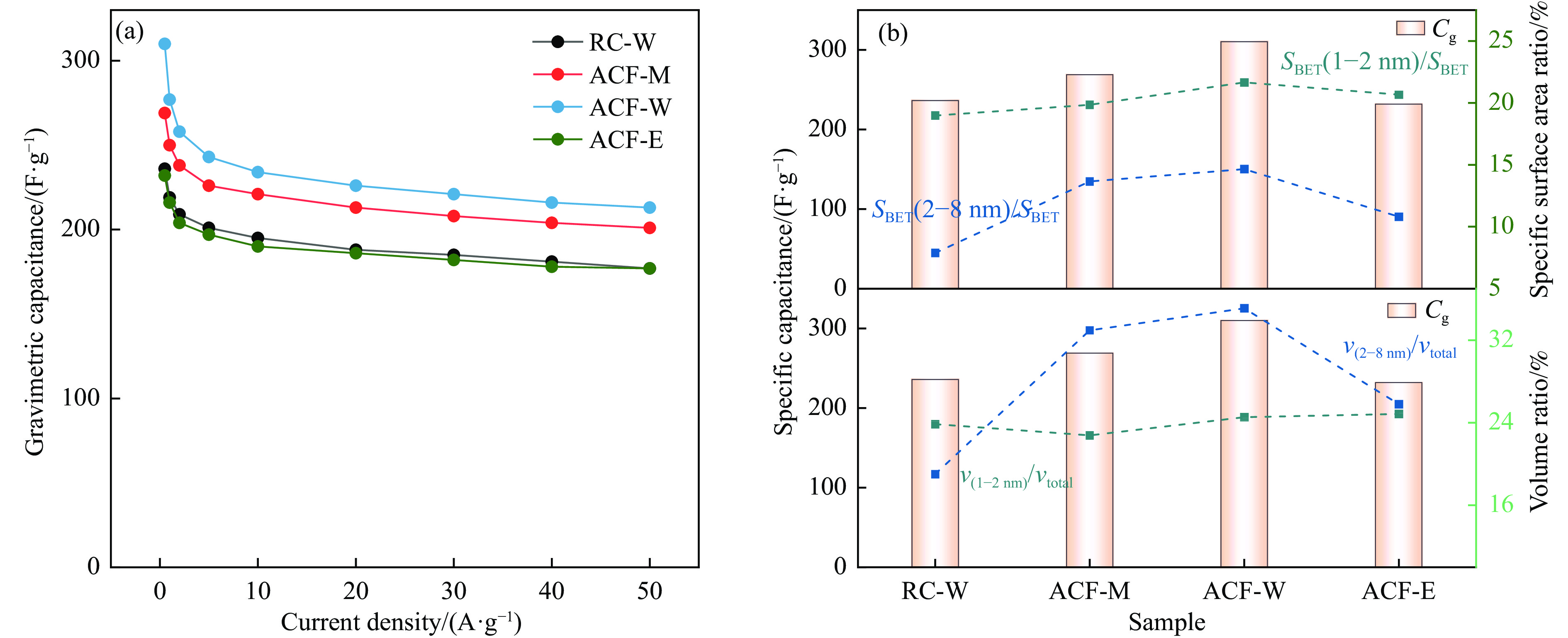
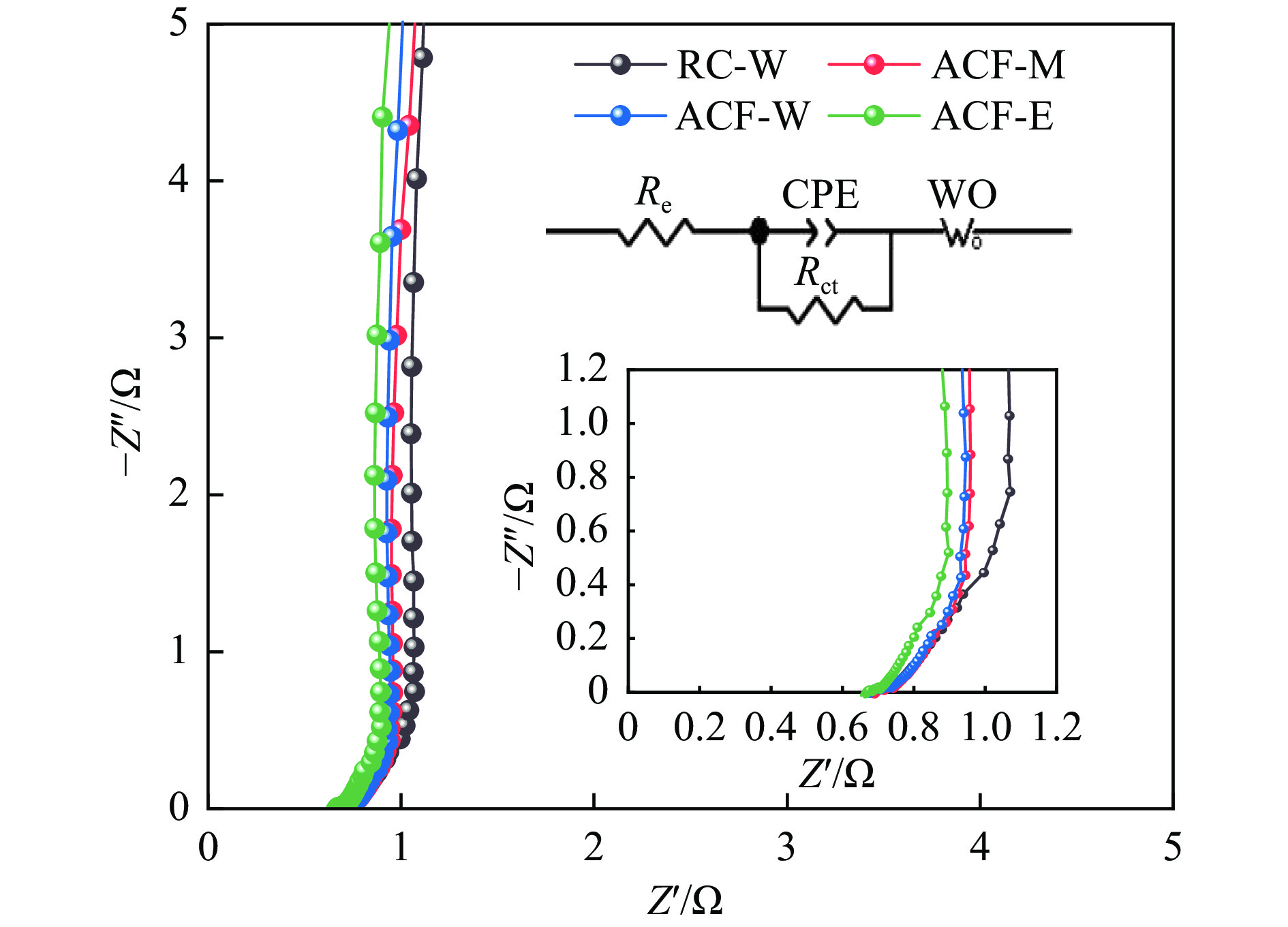
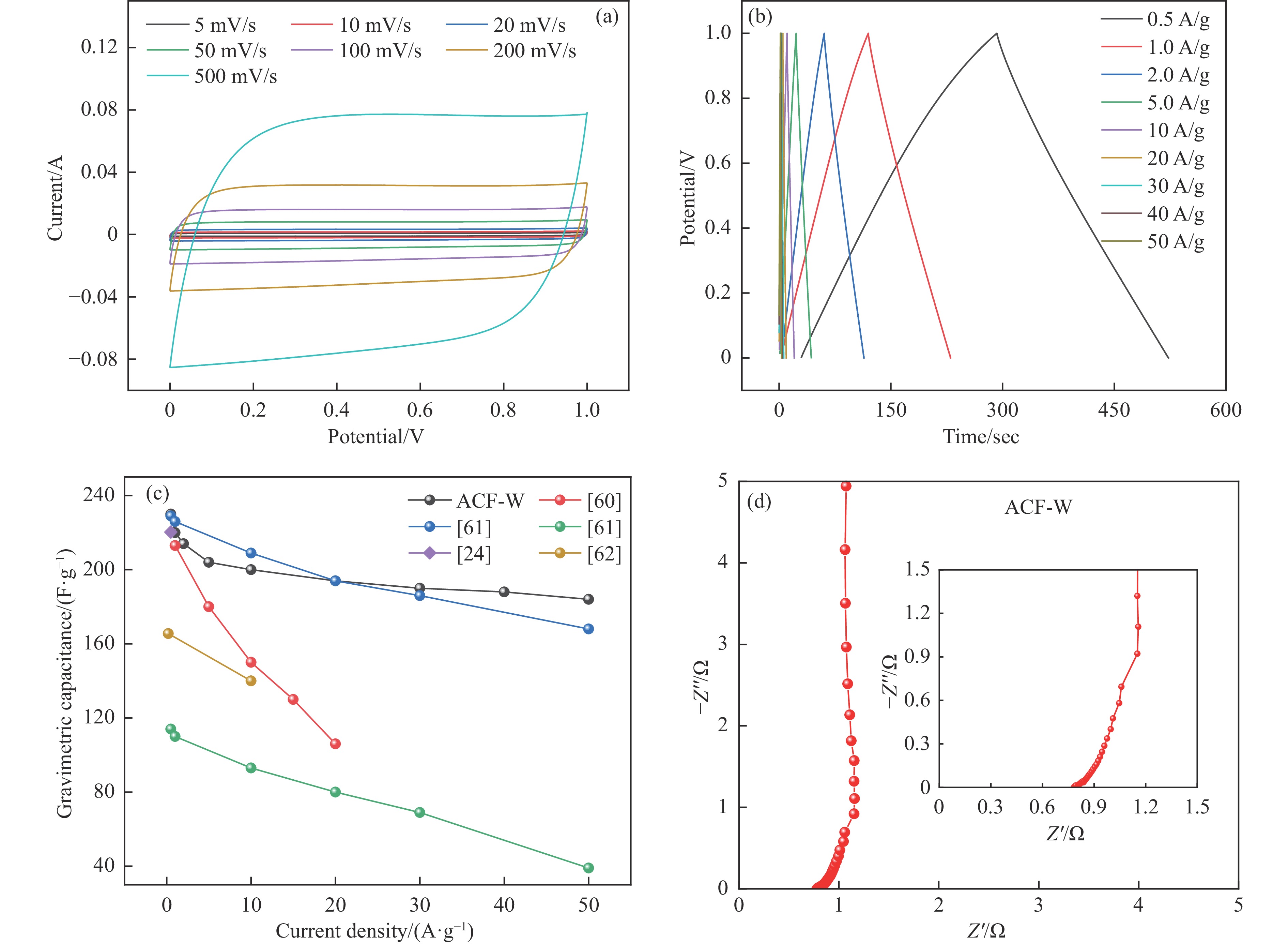
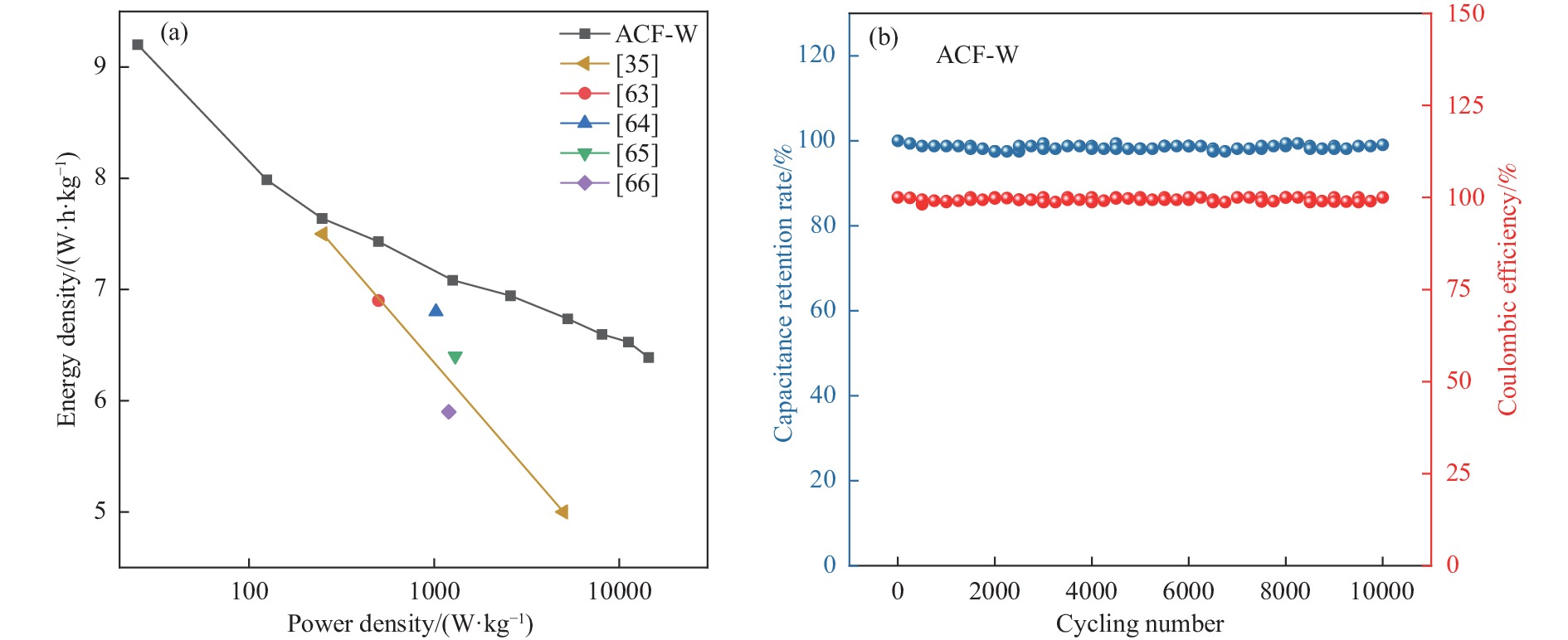
摘要: 以强黏性炼焦煤为原料,经常压自发泡法制得的煤基泡沫炭(NCF)为碳基底,KOH为活化剂,采用机械混合、水溶液浸渍、乙醇浸渍三种不同的方式制备煤基活性泡沫炭(HPCs),并将其用作双电层电容器的电极材料,研究了KOH添加方式对其微观结构和电化学性能的影响。结果表明,KOH分散度和附着性对煤基活性泡沫炭孔隙结构的生成、晶体结构、表面化学性质以及电化学性能有显著影响。煤基泡沫炭本身具有三维连通泡孔结构,有利于活化剂(KOH)深入材料的泡孔内部并为其提供大量附着位点,增大活化剂与碳基体的接触面积进而发生高效的活化。KOH水溶液的流动性较好,可以使K+ 更有效地穿插在NCF的泡孔结构中,在活化过程中作用于缺陷部位,在碳基体内部基质上产生更多的微孔以及介孔结构,有效地放大了活化效果。KOH水溶液浸渍泡沫炭材料制得的ACF-W样品拥有最高的比表面积(3098.35 m2 /g)、总孔体积(1.68 cm3 /g)、介孔体积比(59.13%),将其用作电极材料表现出优异的比电容(310 F/g)以及循环稳定性。
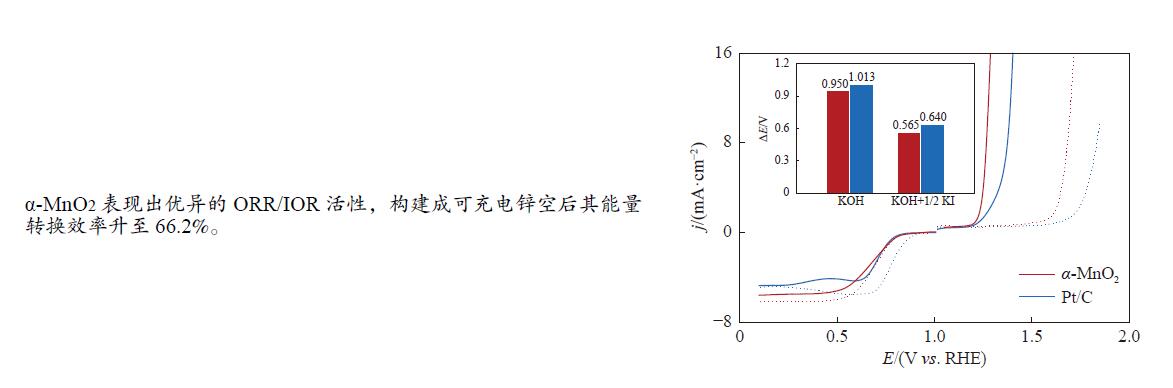
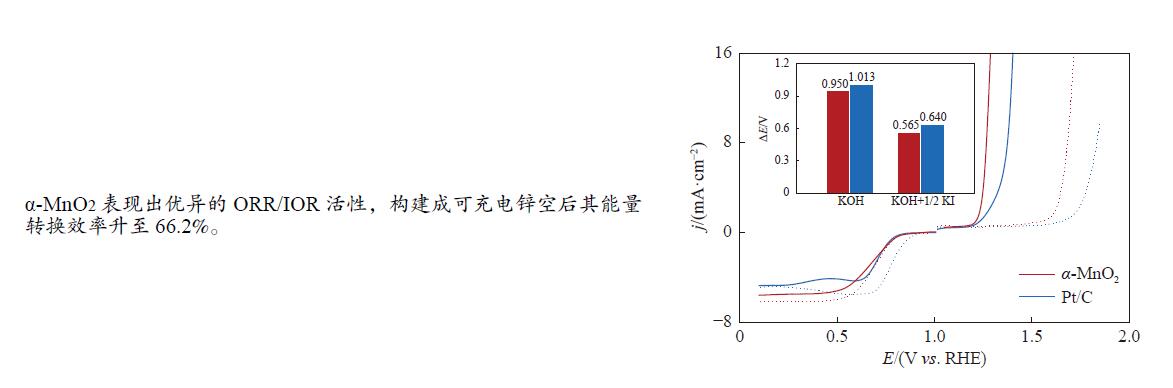
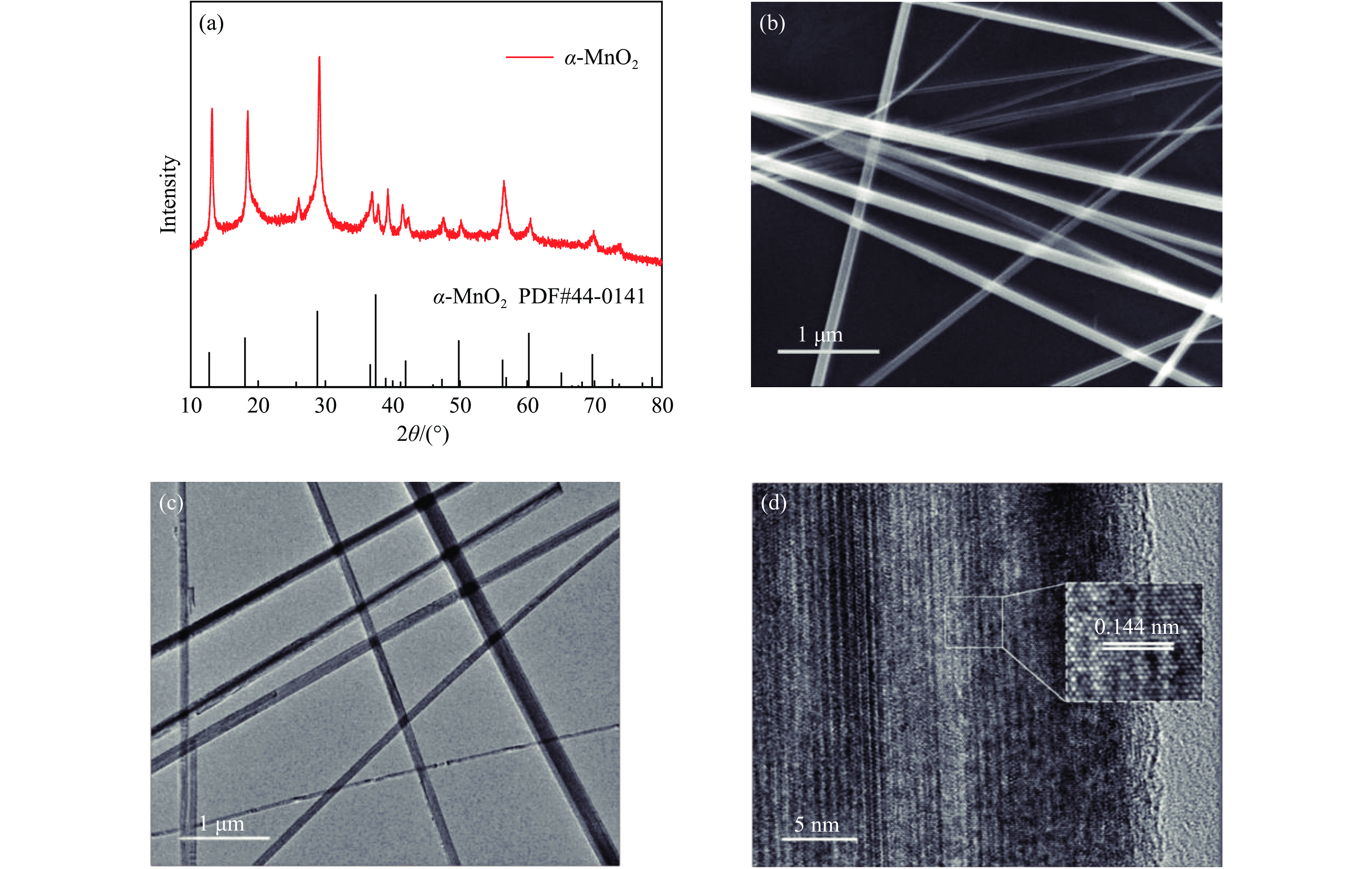
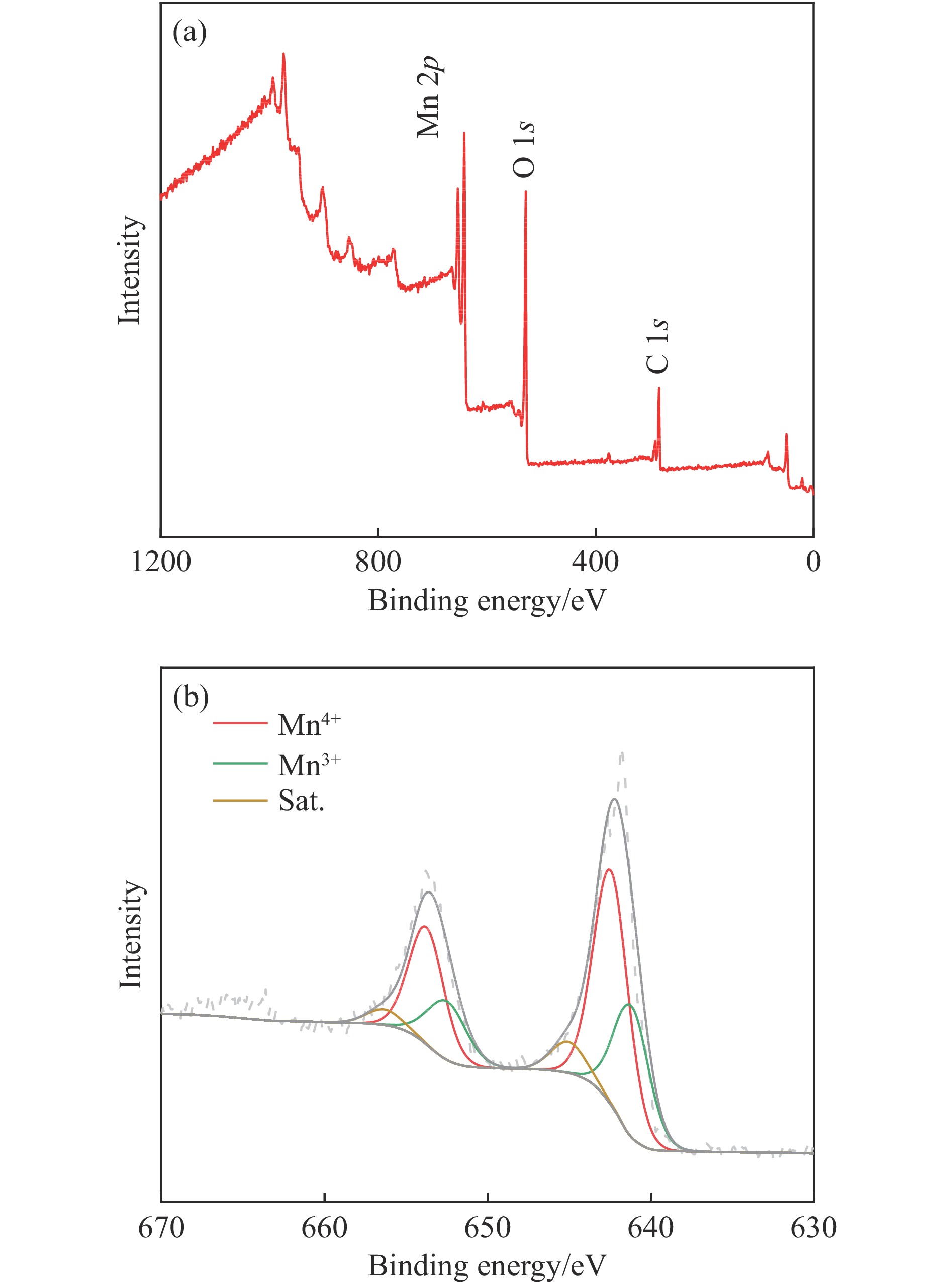
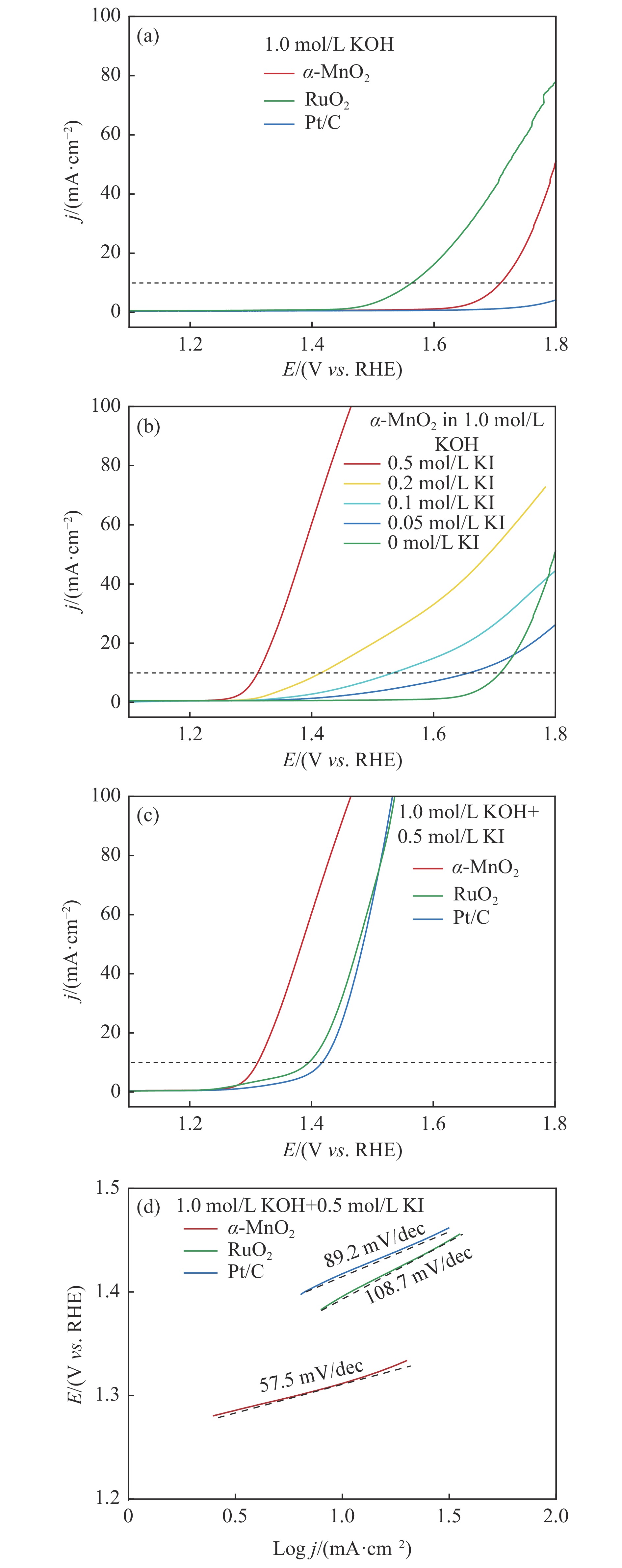
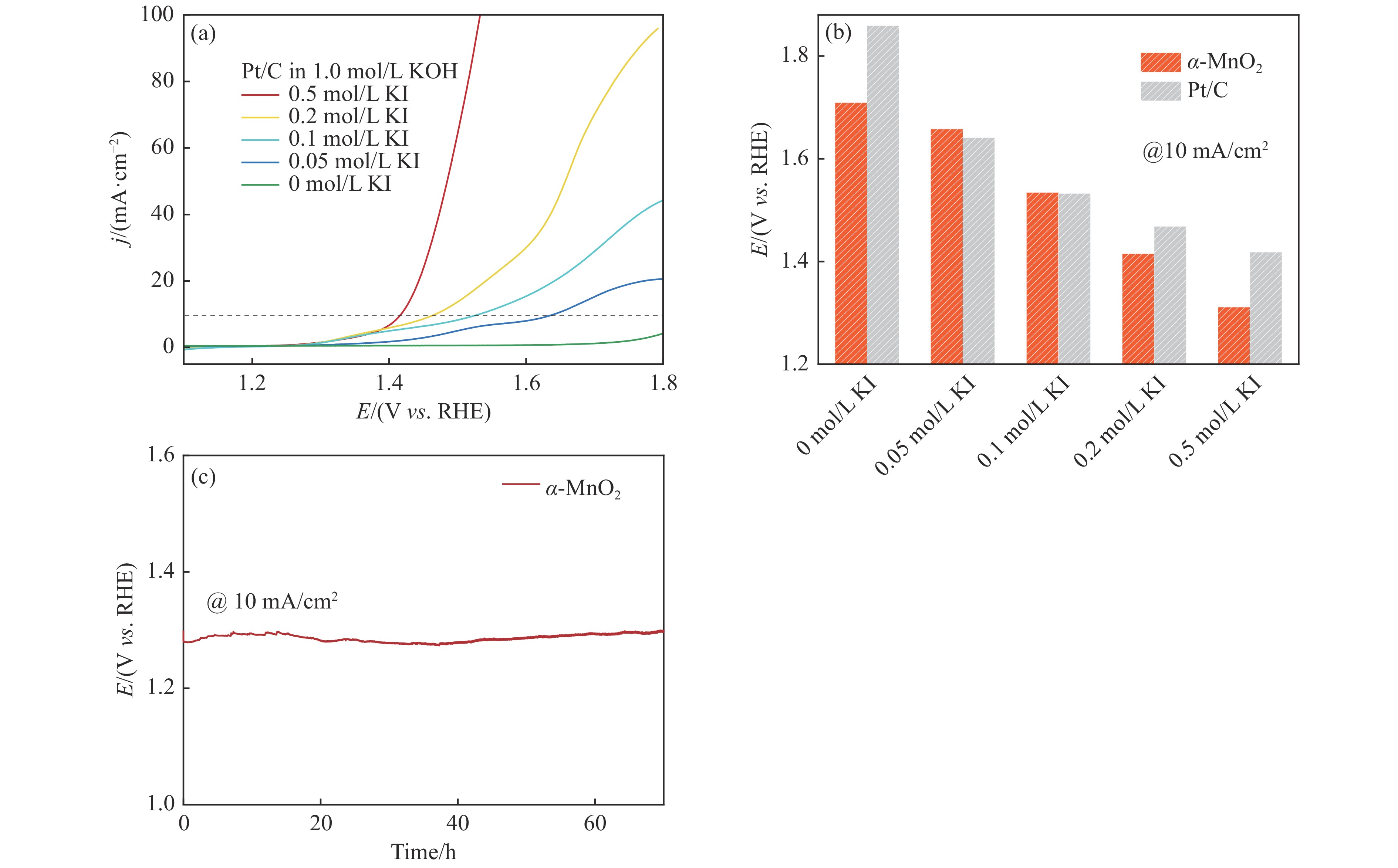
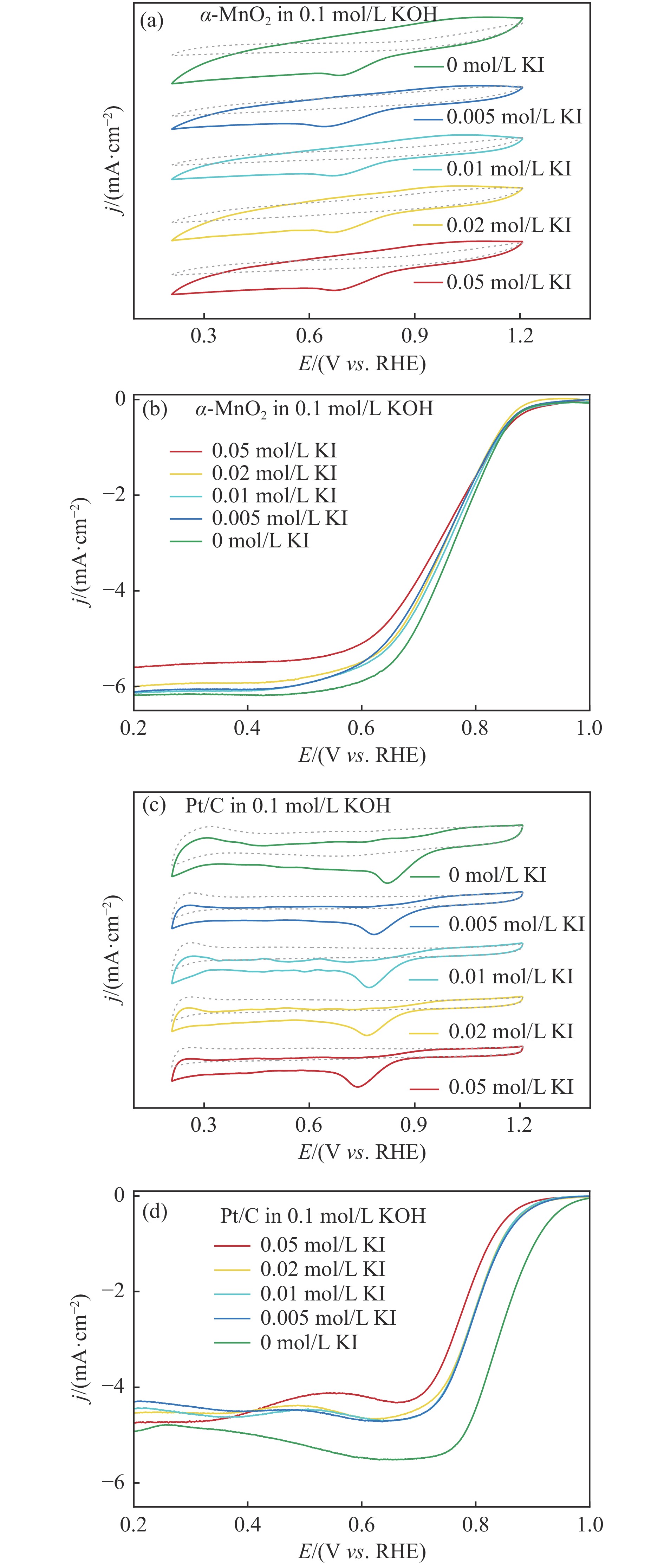
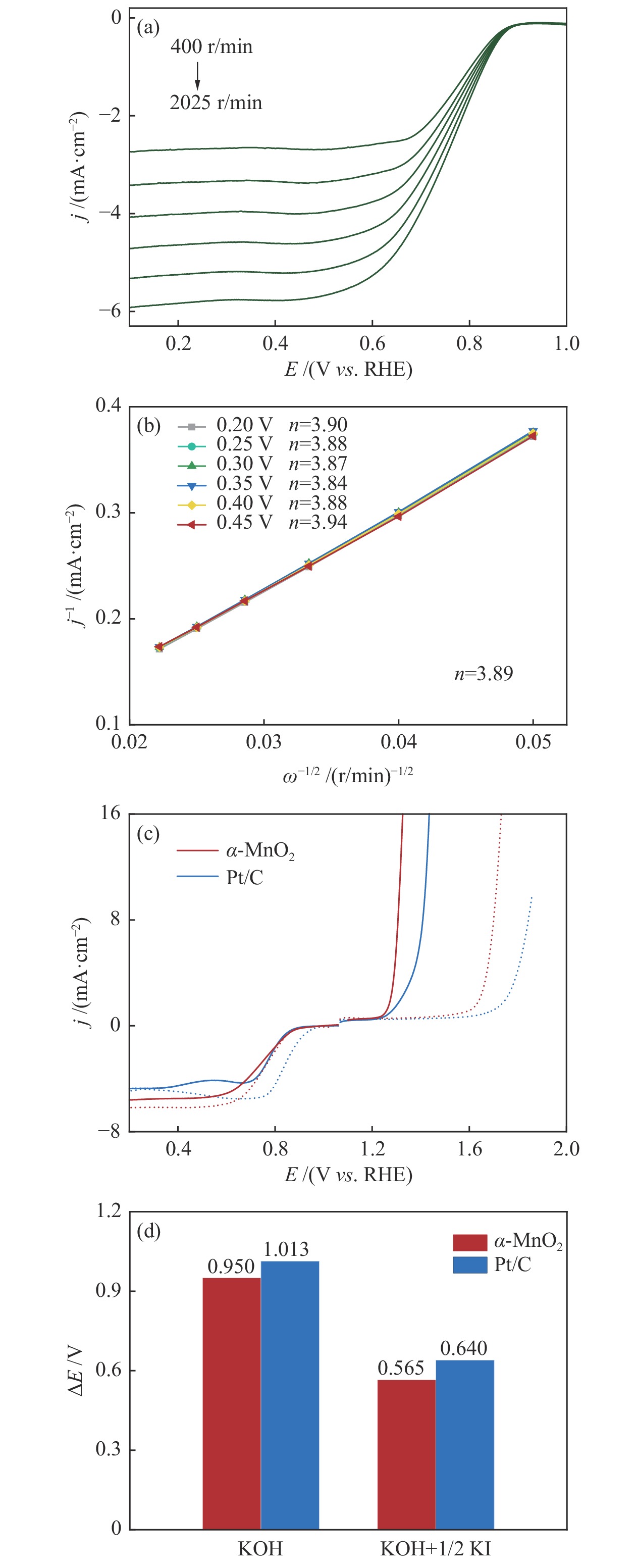
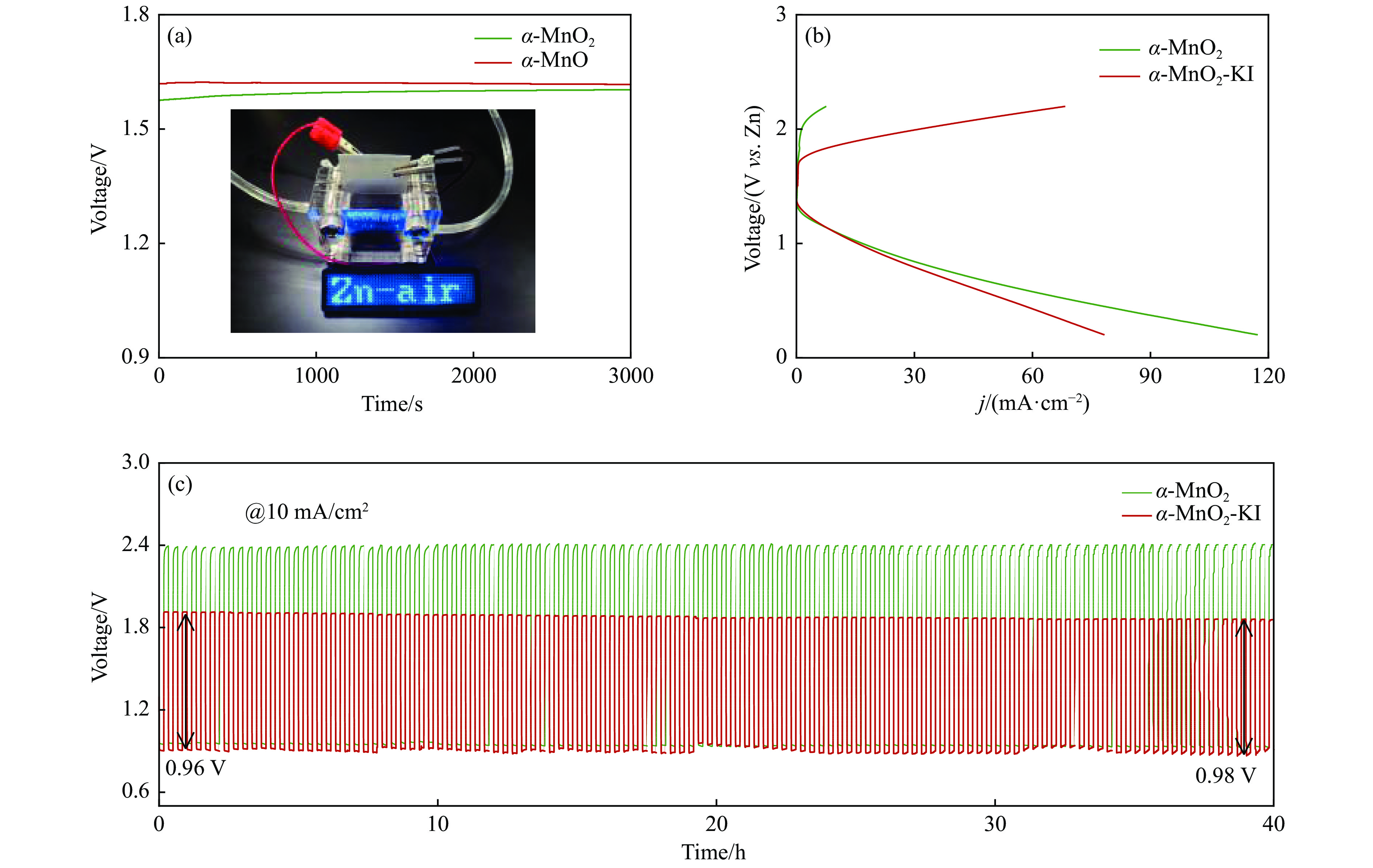
摘要: 析氧反应(oxygen evolution reaction, OER)和氧还原反应(oxygen reduction reaction, ORR)是可充电锌空电池(rechargeable Zn-air batteries, RZABs)重要的两个反应。其中,析氧反应具有较高的热力学平衡电位和复杂的反应路径,实际应用中需要高的充电电压驱动其发生,这将带来一系列问题并且限制了RZABs的商业化应用。基于此,本研究构造α -MnO2 并作为ORR/IOR双功能催化剂。在碱性体系中引入反应改性剂KI,α -MnO2 对碘离子氧化反应(iodide oxidation reaction, IOR)具有更低的阳极氧化电位和更快的催化动力学。当1.0 mol/L KOH电解液中添加0.5 mol/L KI时,相比于OER(1.709 V @10 mA/cm2 ),α -MnO2 在IOR过程中电流密度达到10 mA/cm2 时阳极电位减小了398 mV(1.311 V vs . RHE),且表现出低至57.5 mV/dec塔菲尔斜率。相对于与Pt/C,在含有KI的KOH电解液中,α -MnO2 表现出与Pt/C相媲美的ORR活性。此外,以α -MnO2 为空气电极组装成RZAB后,该电池也表现出了优异的充电活性和良好的循环寿命,在5 mA/cm2 电流密度下,充放电电压间隙由0.97 V缩减为0.61 V,能量转换效率由54.9%提升至66.2%。
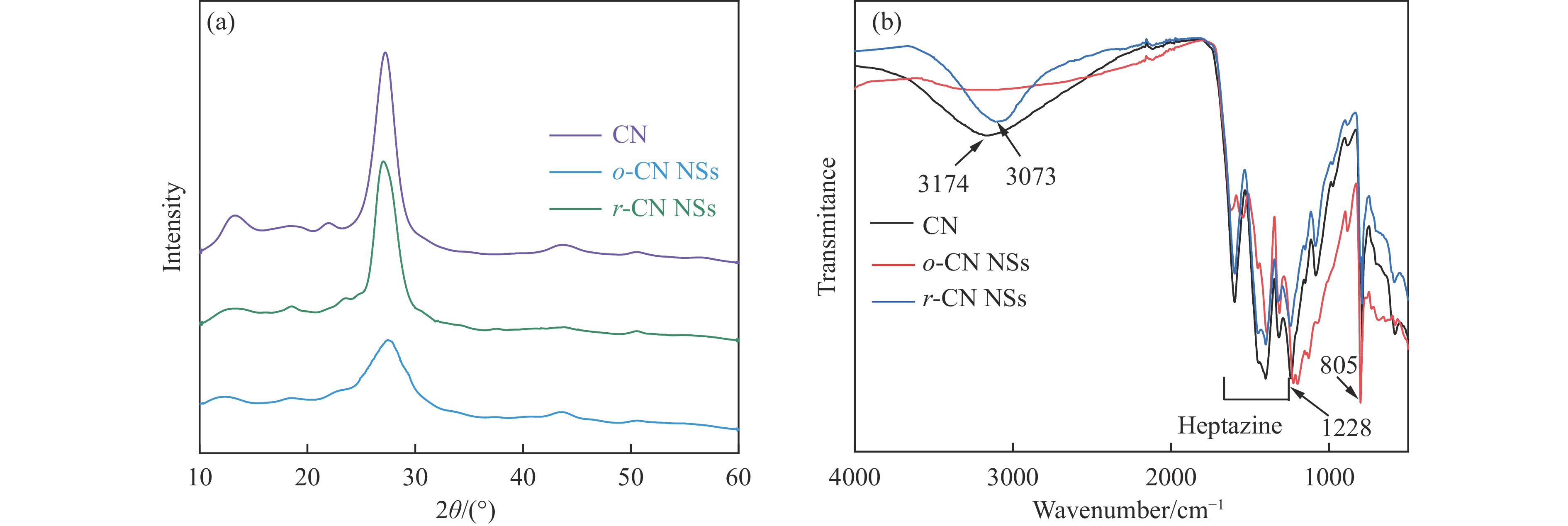
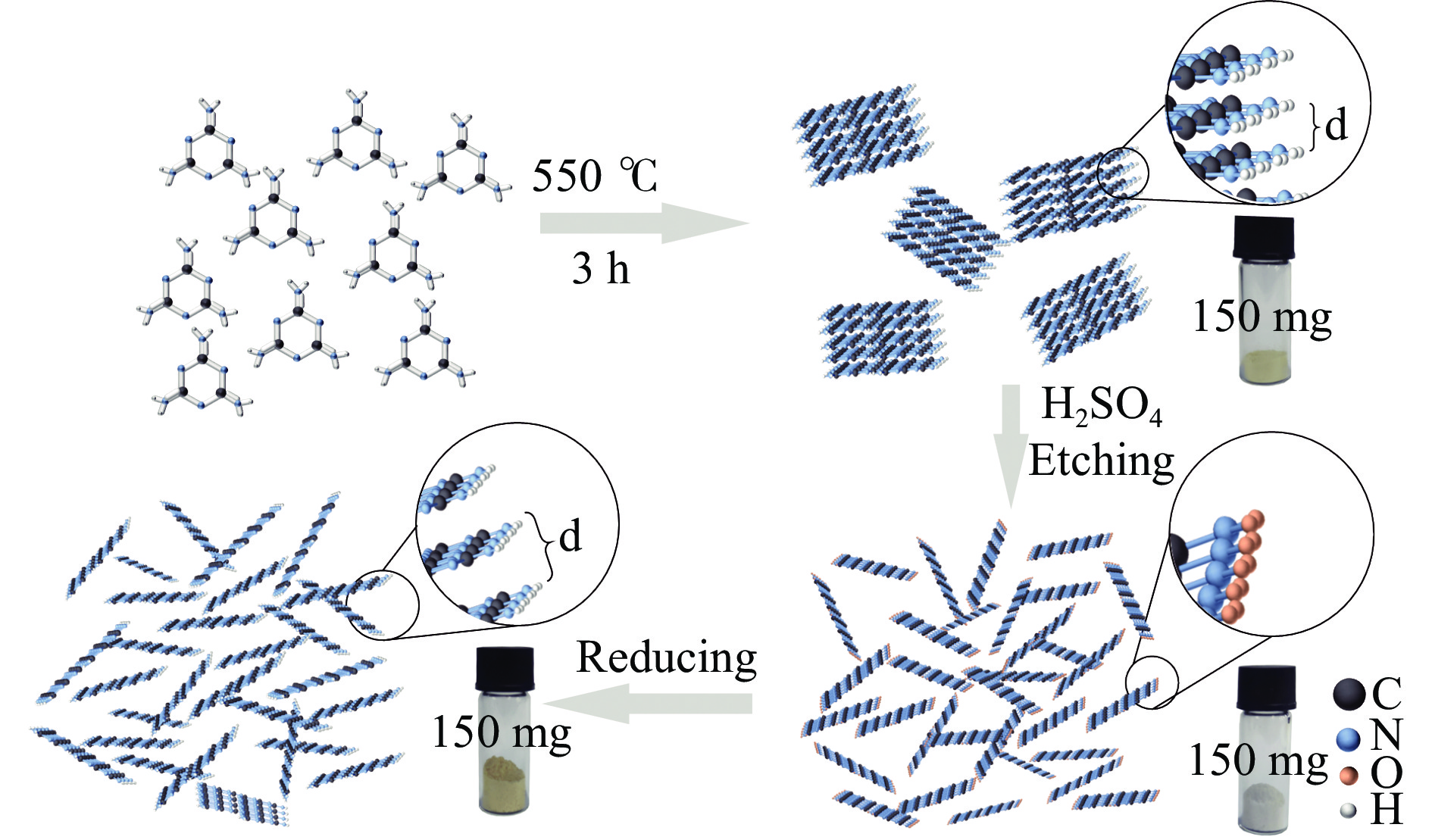
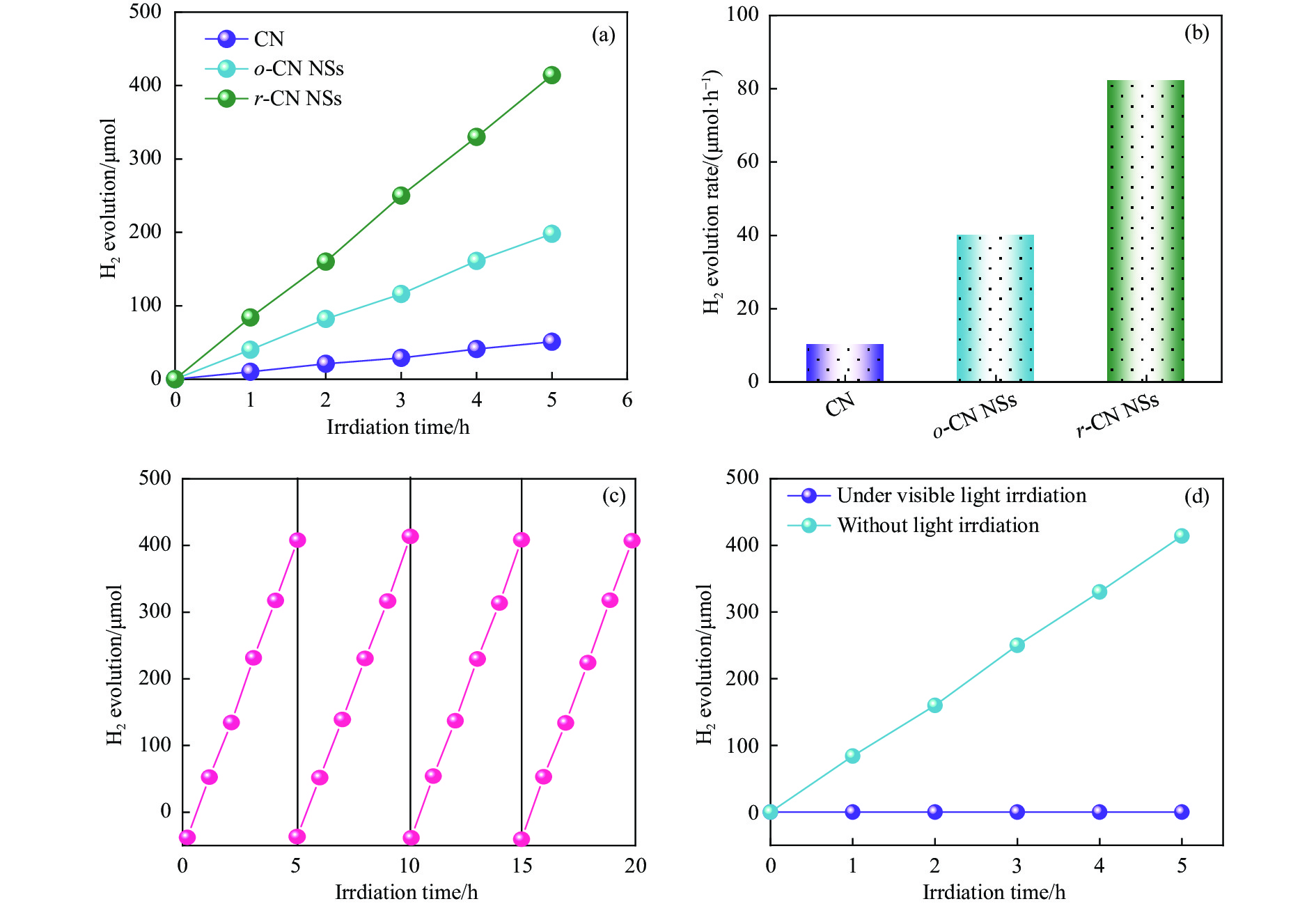
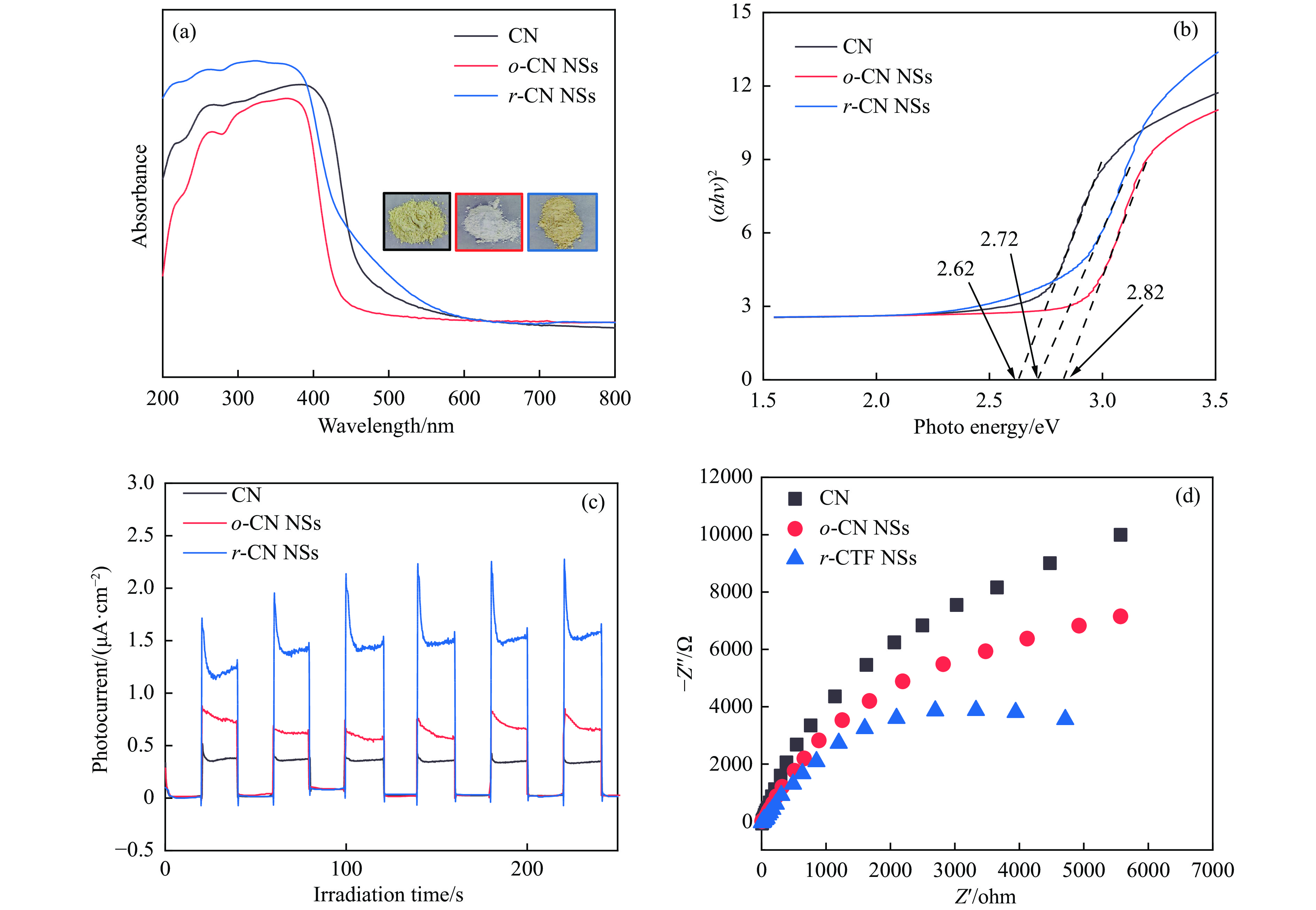
摘要: 以块体氮化碳 (CN) 为前驱物,采用氧化剥离制备氧化型氮化碳纳米片 (o -CN NSs),将o -CN NSs还原制得了还原型氮化碳纳米片 (r -CN NSs)。o -CN NSs和r -CN NSs厚度均约2 nm,且都保留了纯CN的庚嗪环骨架结构;相比于o -CN NSs,r -CN NSs具有更小的禁带宽度 (2.62 eV)、更宽的光响应范围 (485 nm) 和更高的产氢速率((1700 μmol/(g·h));r -CN NSs的光催化产氢速率是CN的8.5倍、o -CN NSs的2.1倍。经过20 h的循环测试,r -CN NSs的光催化产氢速率没有衰减,具备良好的光催化稳定性。实验和理论分析表明,r -CN NSs是边缘基团为氨基的纳米片结构,氨基的引入改善了纳米片的结晶性,提高了电子和空穴的分离效率、拓宽了纳米片的光响应范围,从而导致光催化性能增强。
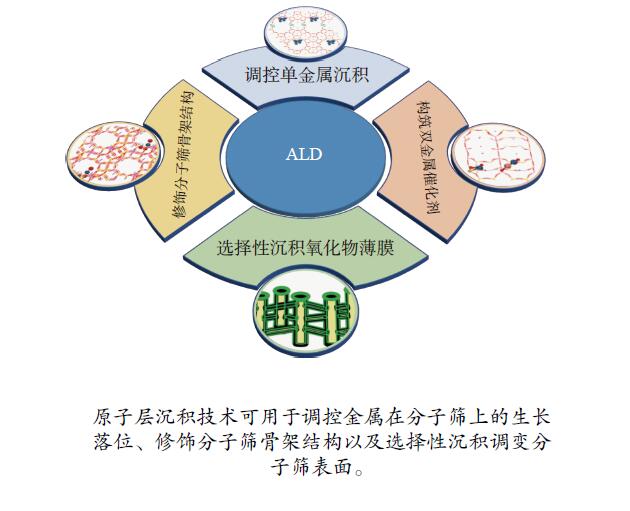
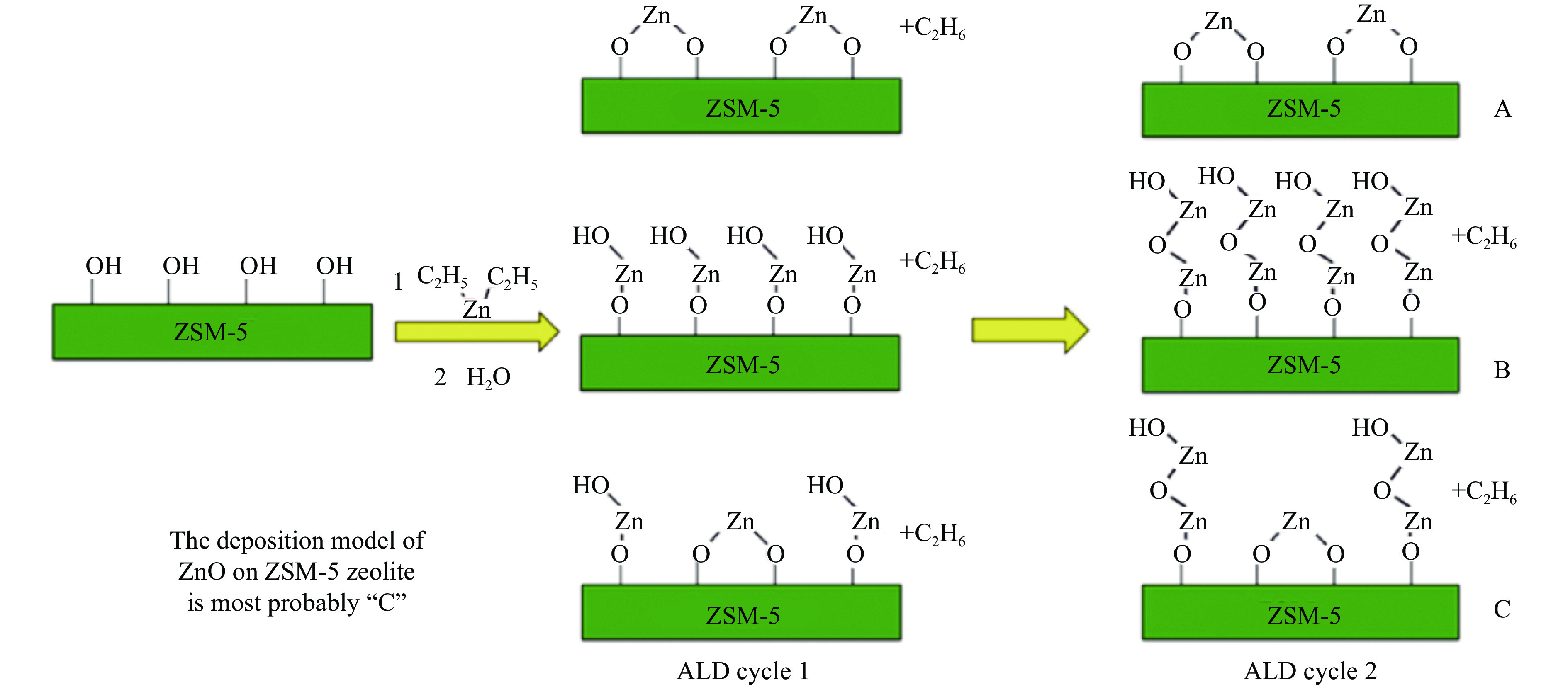
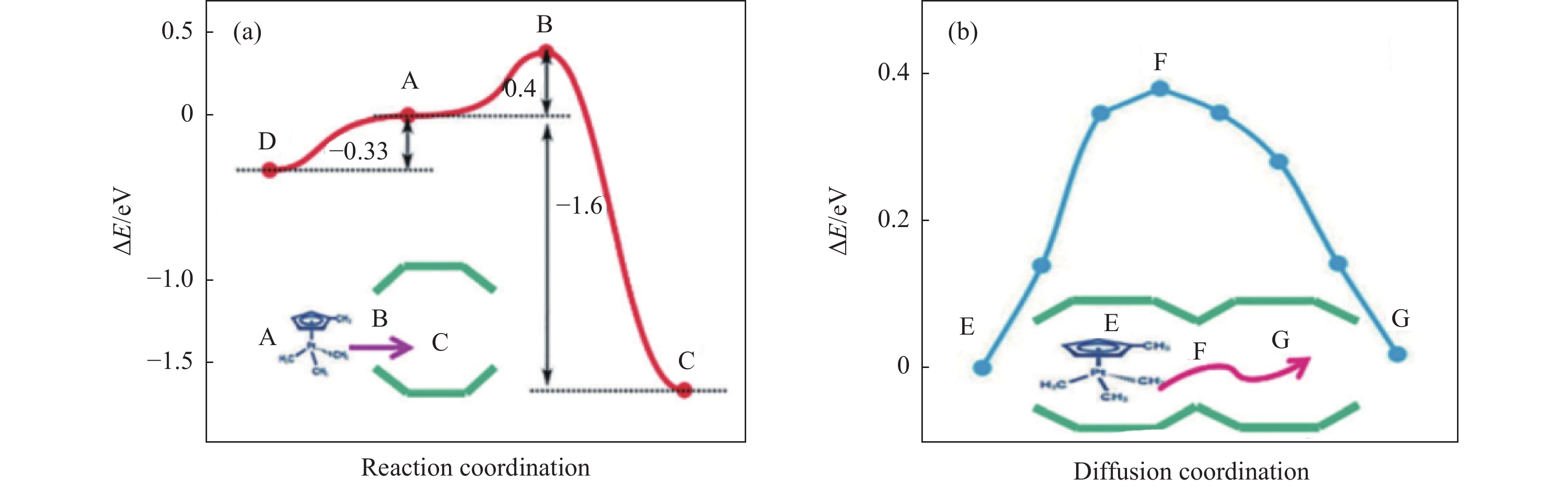
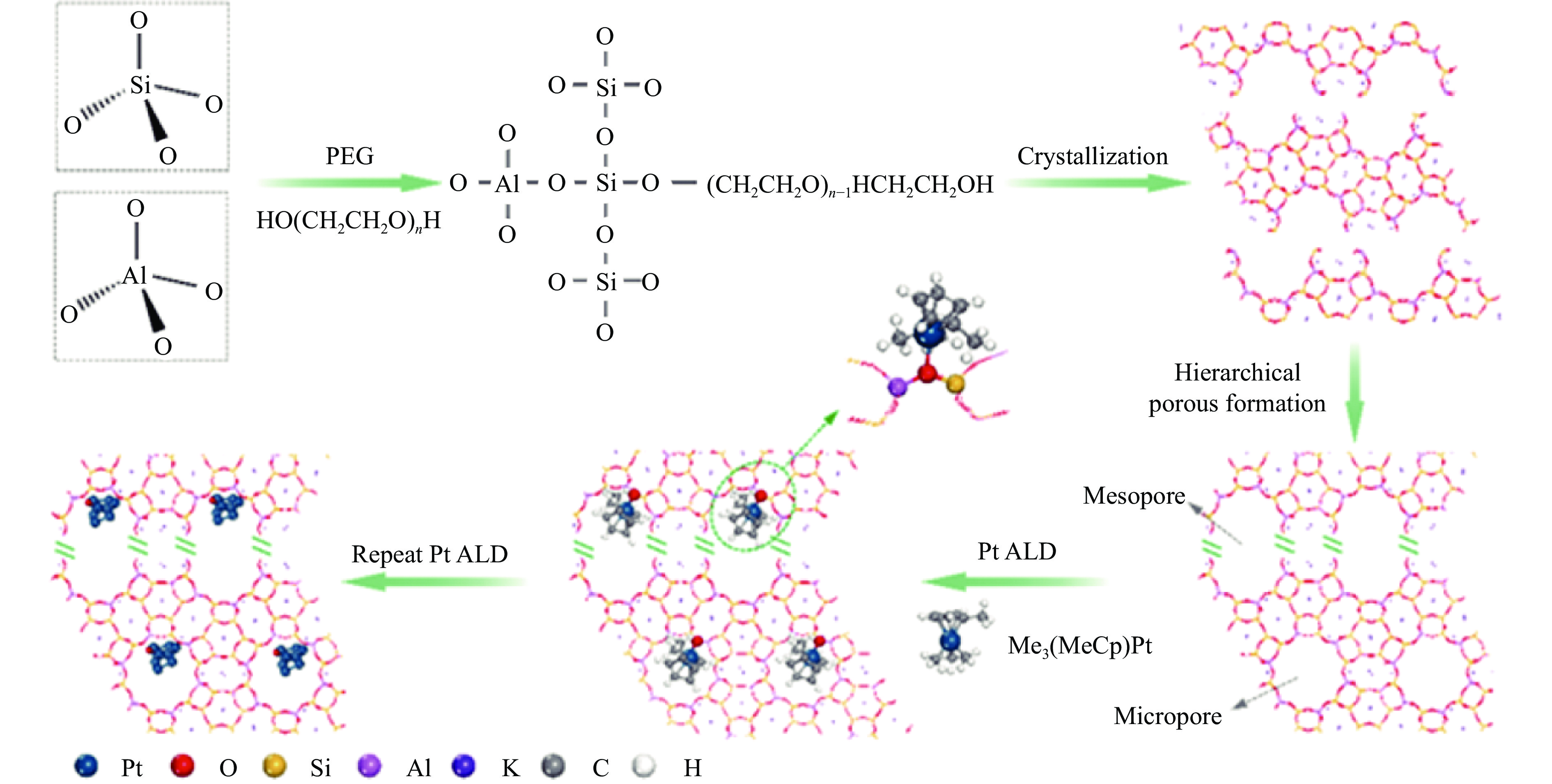
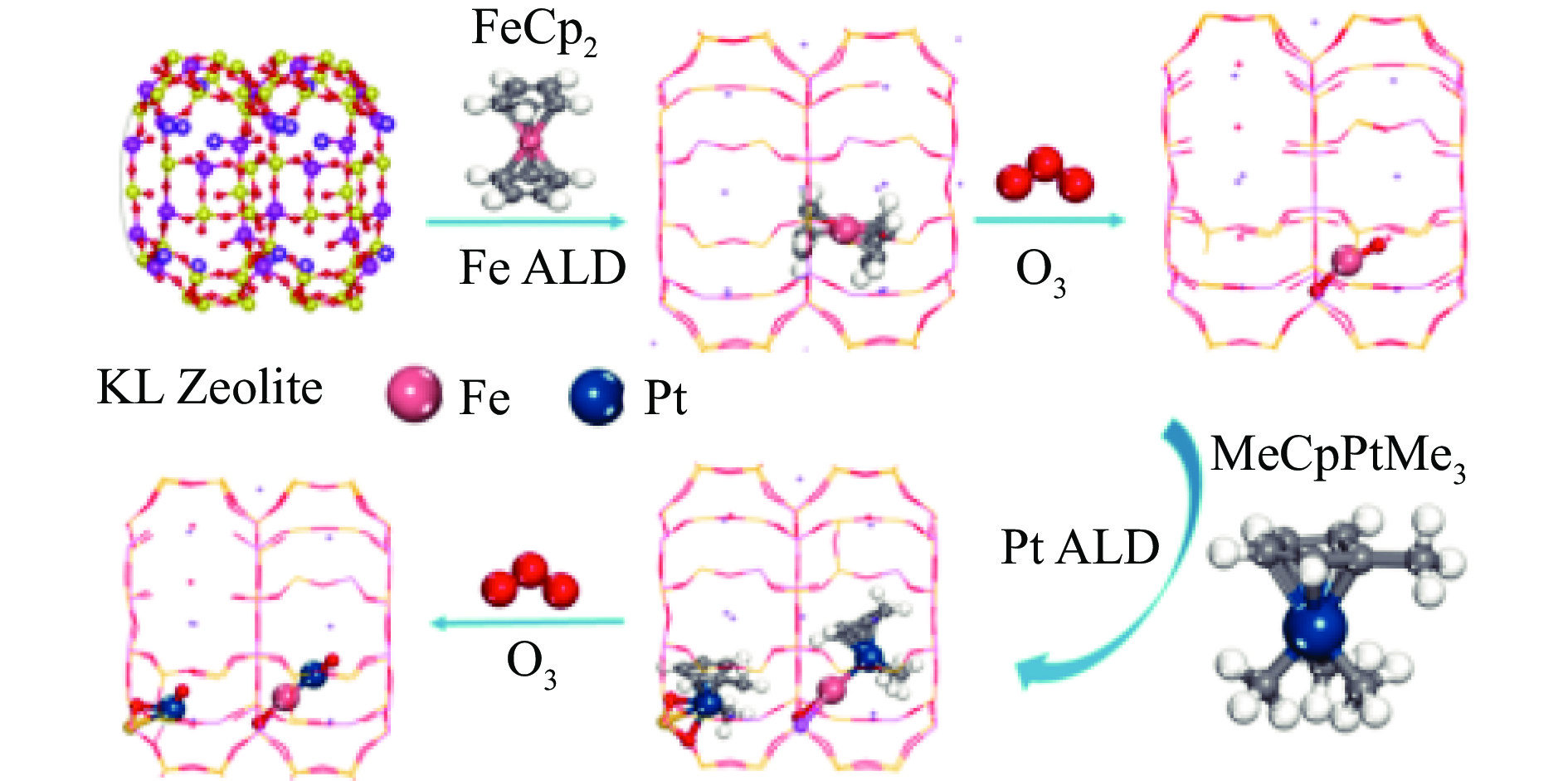
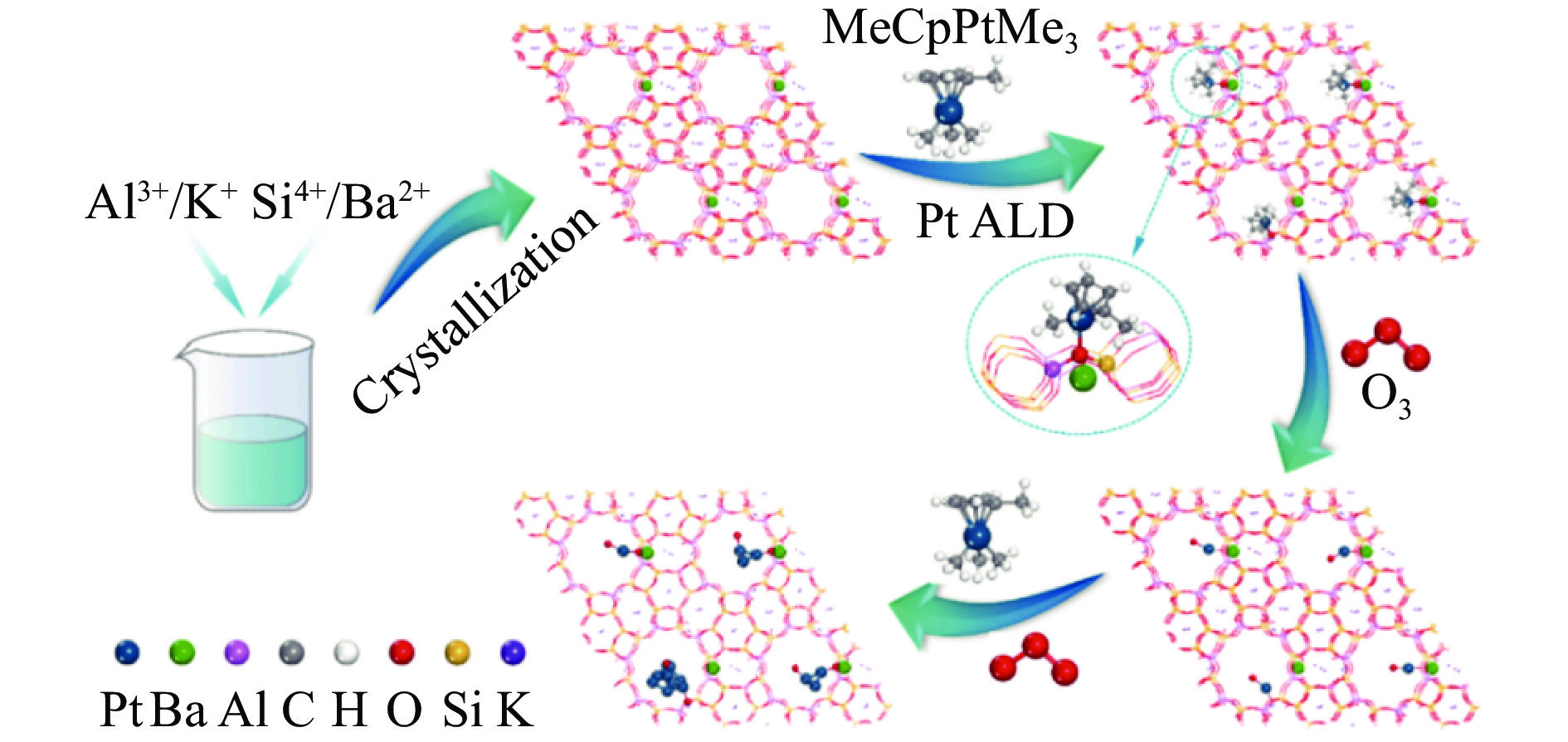
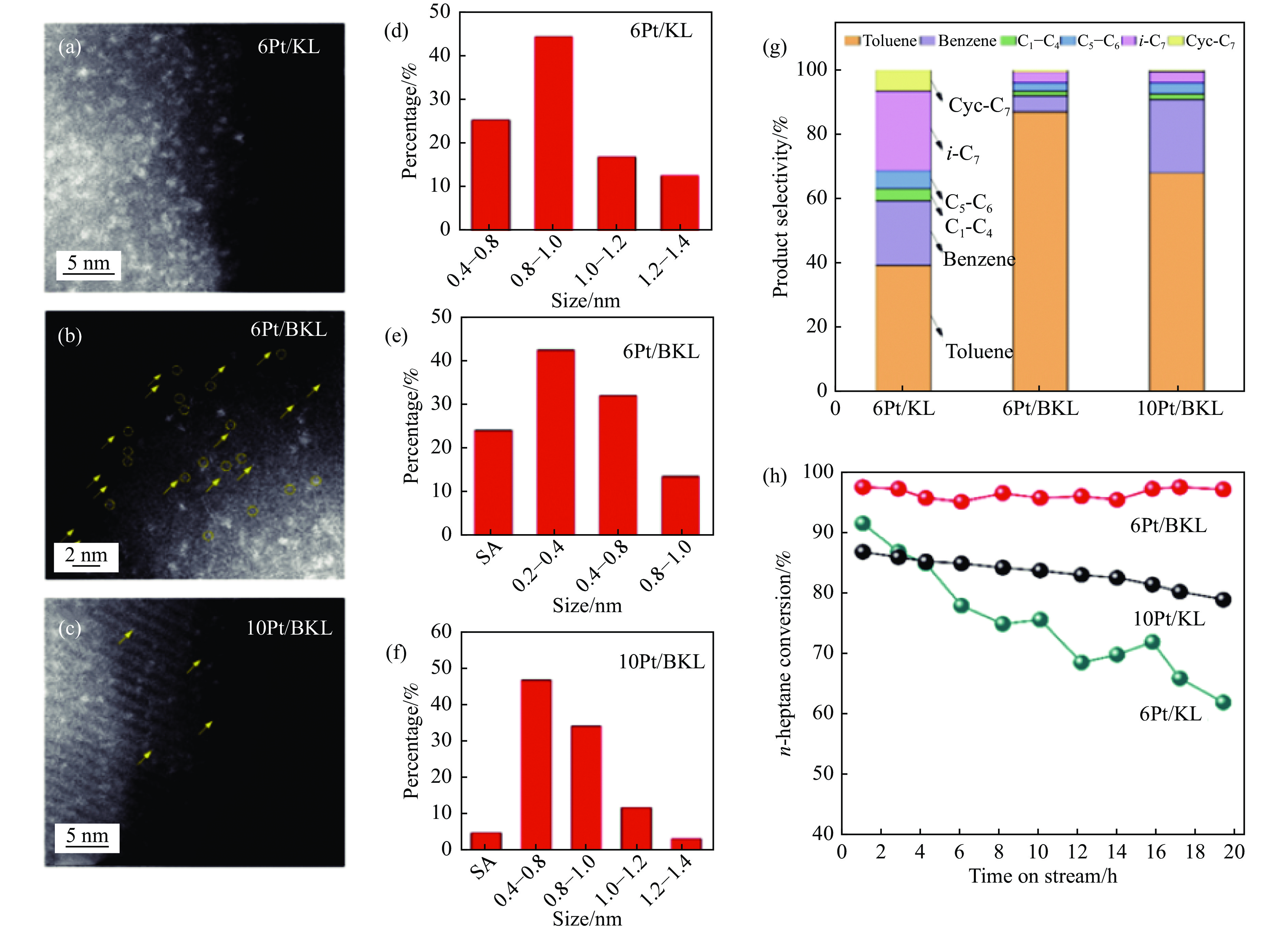
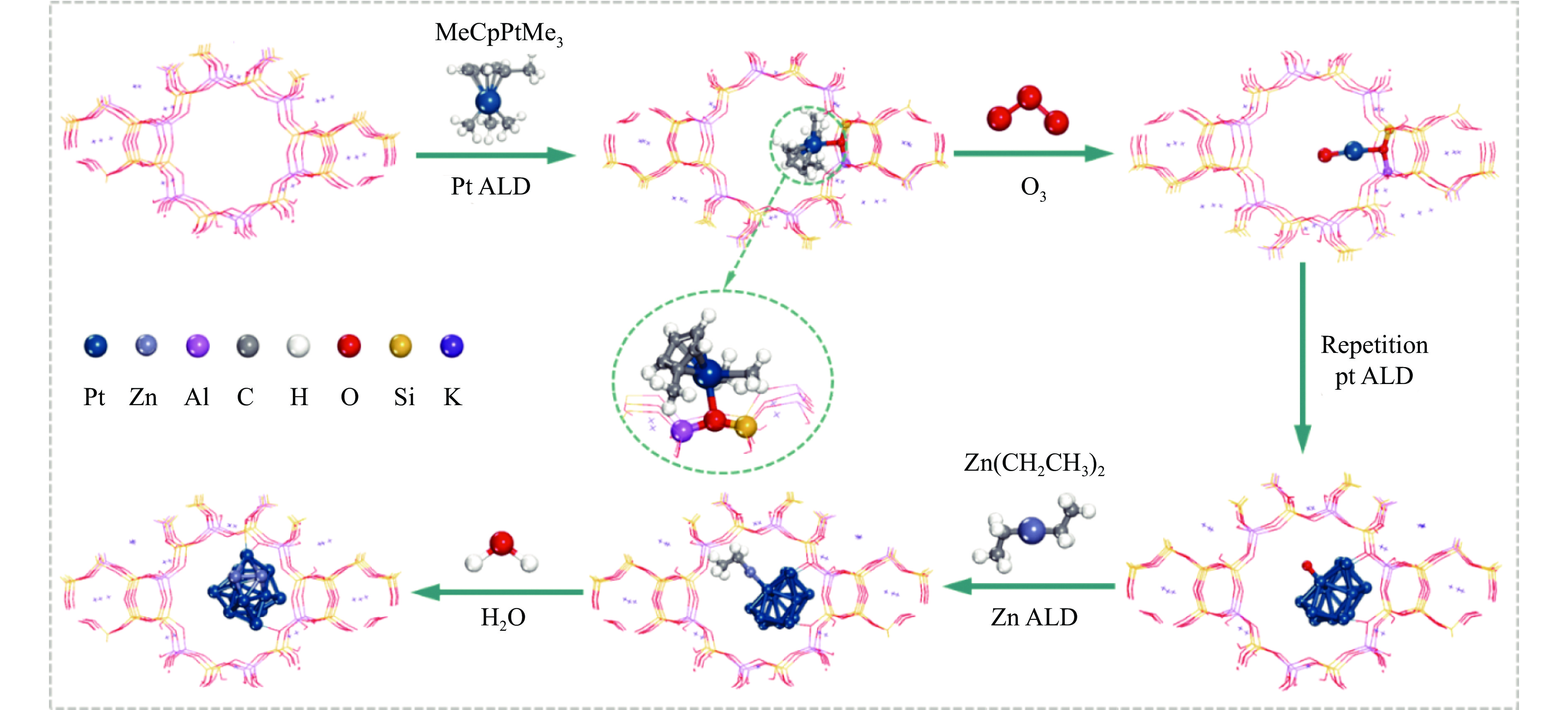
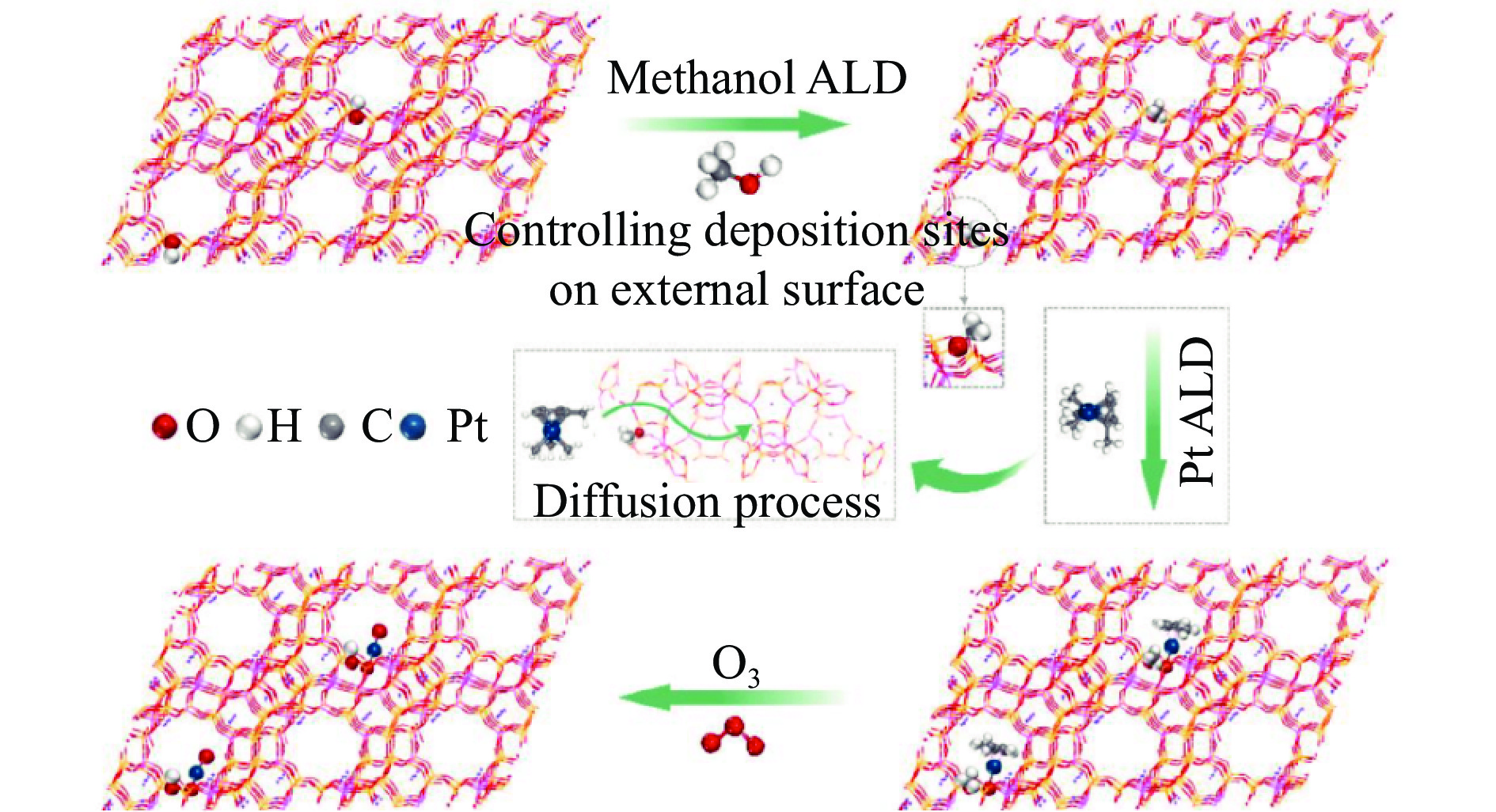
摘要: 分子筛基催化剂在多相催化研究领域具有重要的应用,但调控活性中心粒子的结构及其在分子筛上的空间位置仍比较困难,是科研界和工业界共同面临的巨大挑战。原子层沉积(ALD)是一种先进的薄膜沉积技术,利用其自限制生长优势,可在原子级别实现对金属粒子生长过程的精准调控。本工作综述了ALD技术在制备分子筛基催化剂方面的应用,主要包括利用ALD技术控制活性位点在分子筛上的生长落位、修饰分子筛骨架结构以及选择性沉积膜调变分子筛表面结构。利用ALD技术设计和调控活性组分结构促进了分子筛基催化剂的发展,但由于分子筛孔道结构复杂且存在缺陷位,因此,ALD技术在分子筛基催化剂的设计调控及大规模应用方面仍具有挑战性,也是今后研究工作的重点。