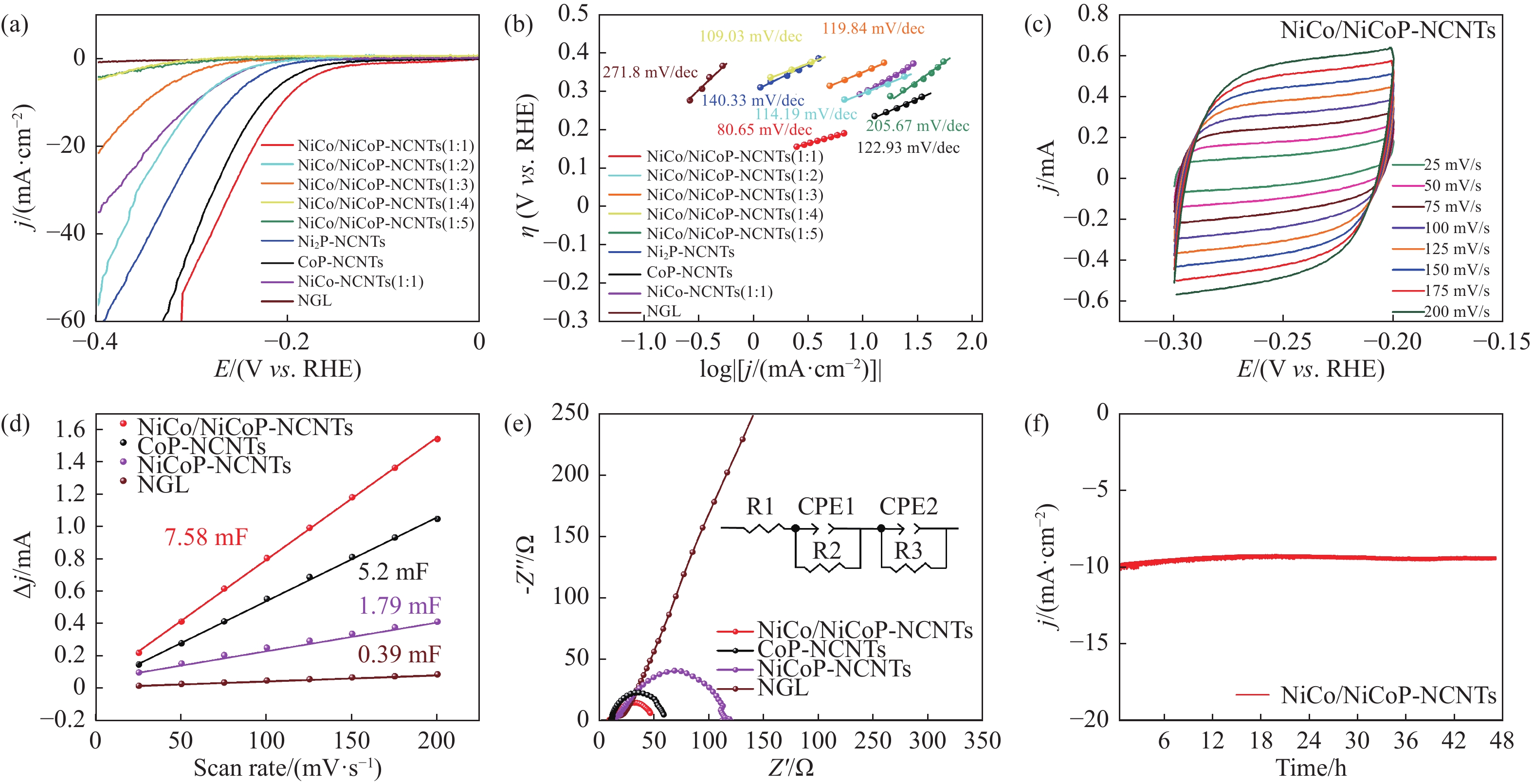
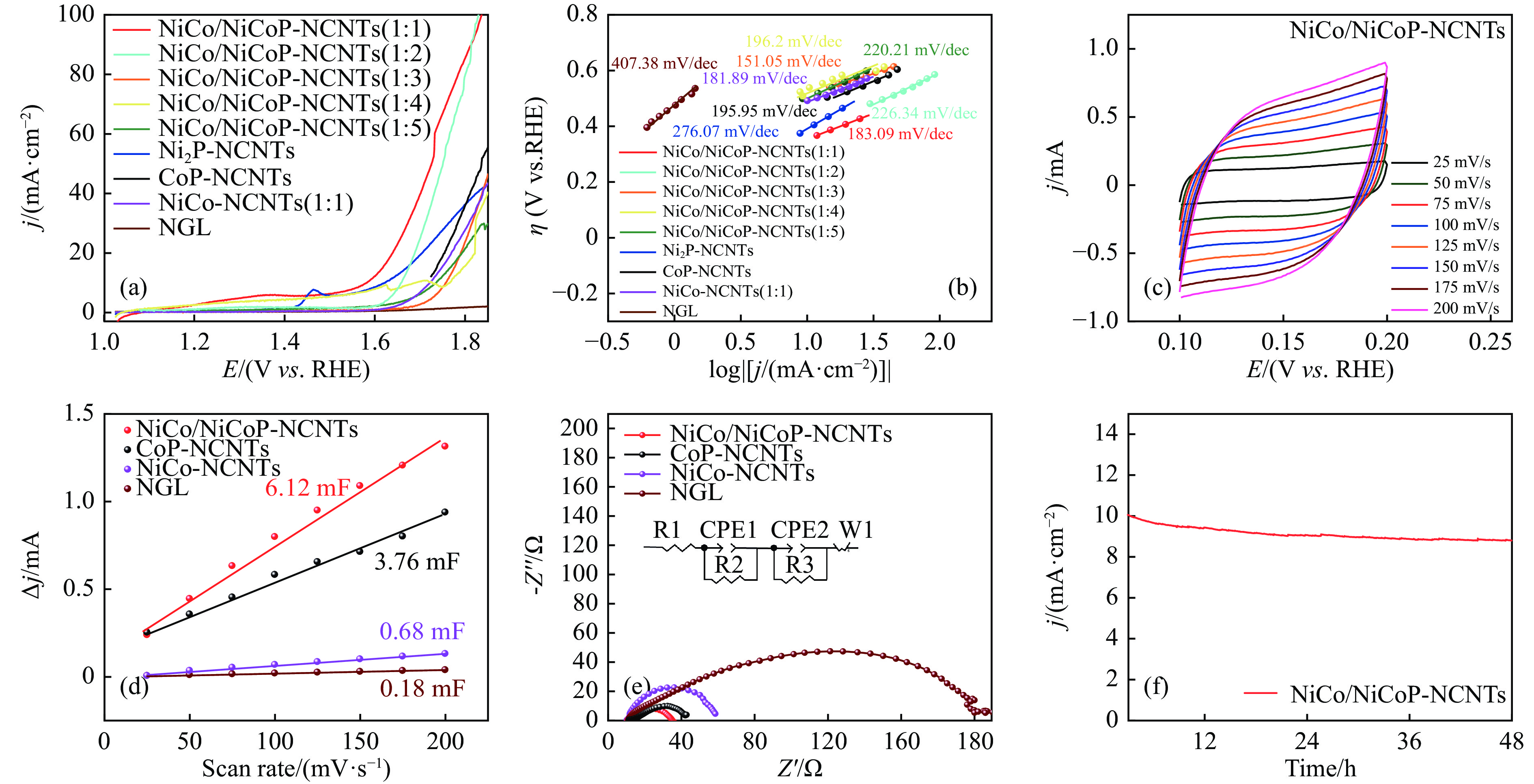
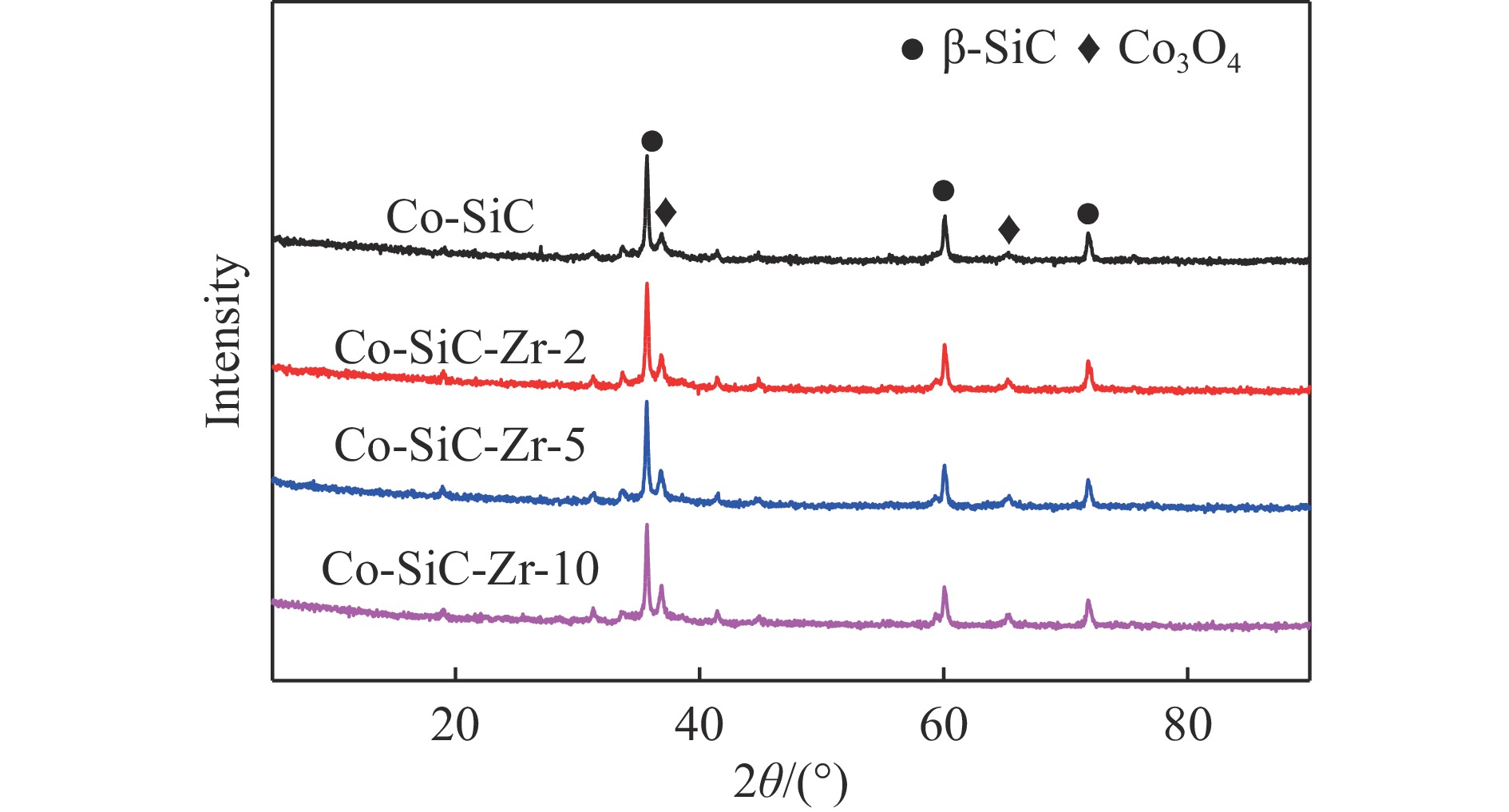
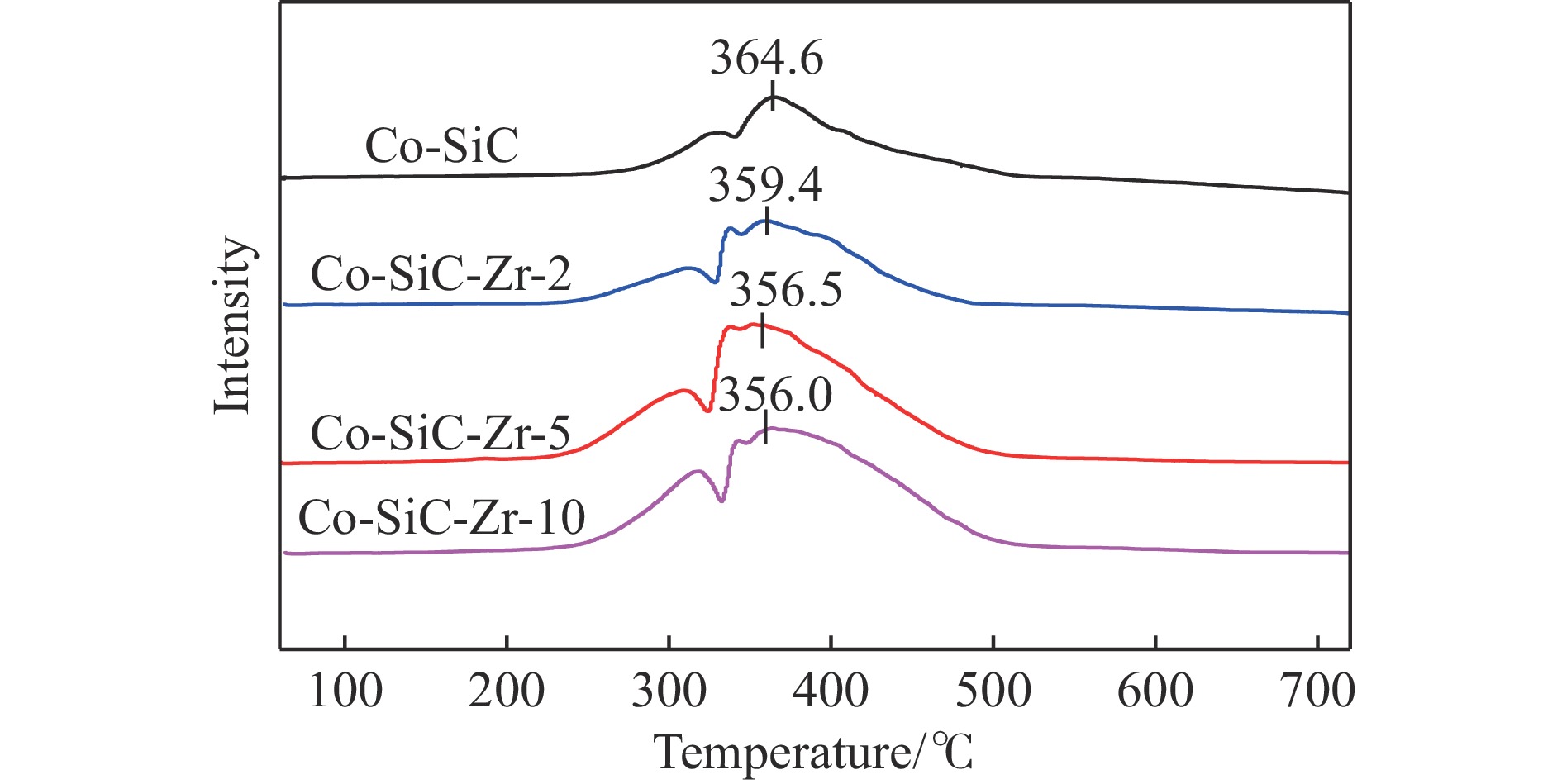
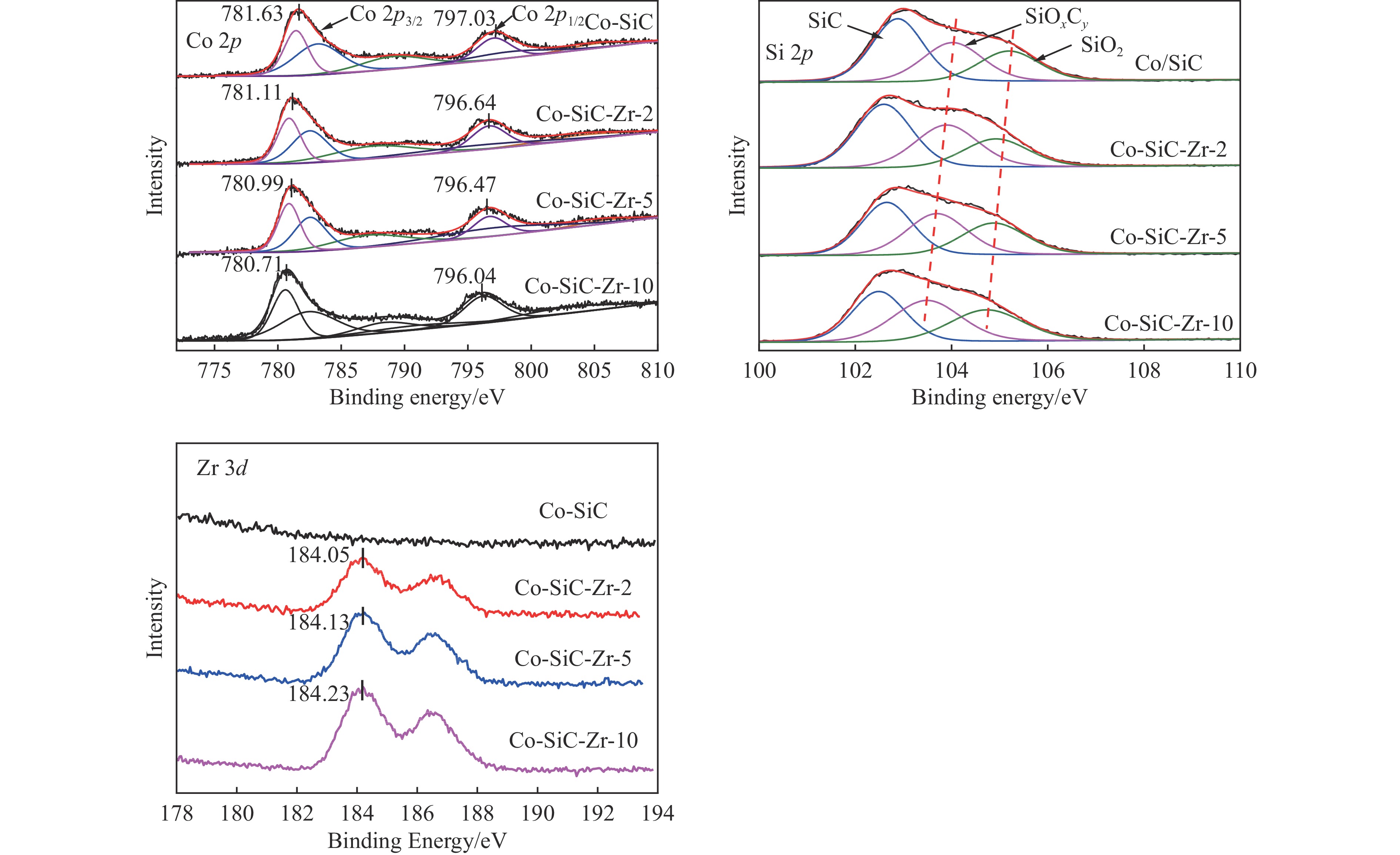
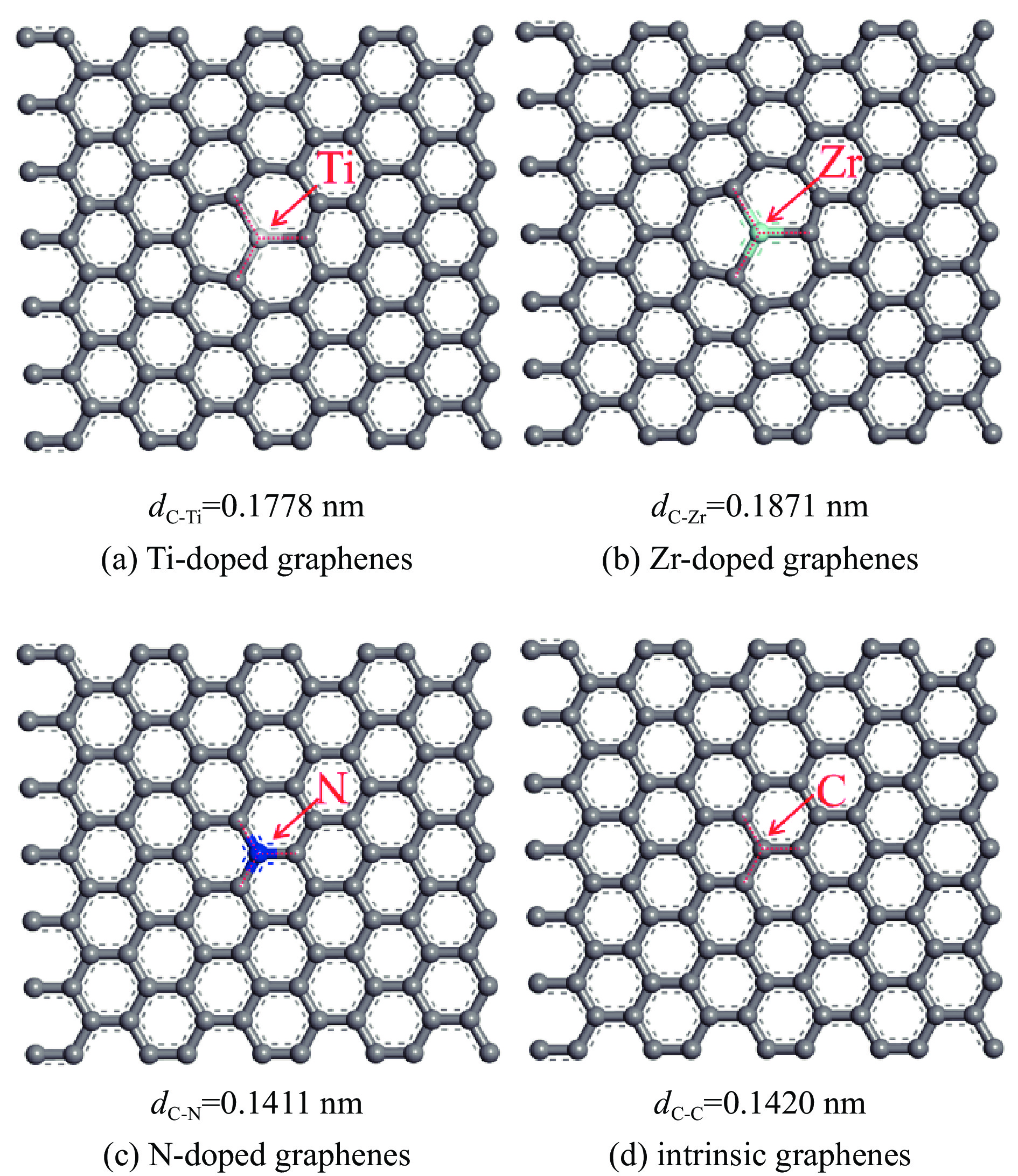
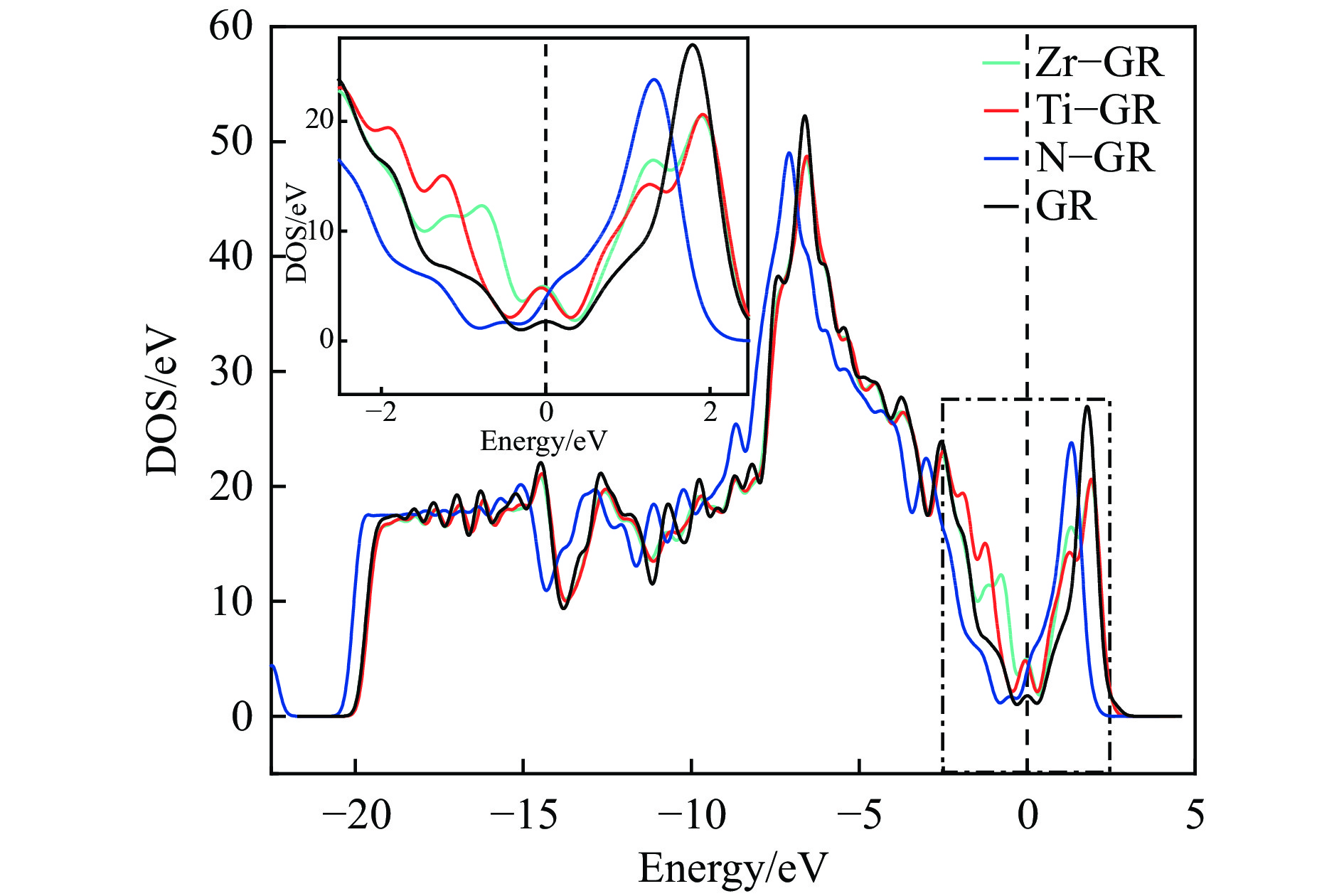
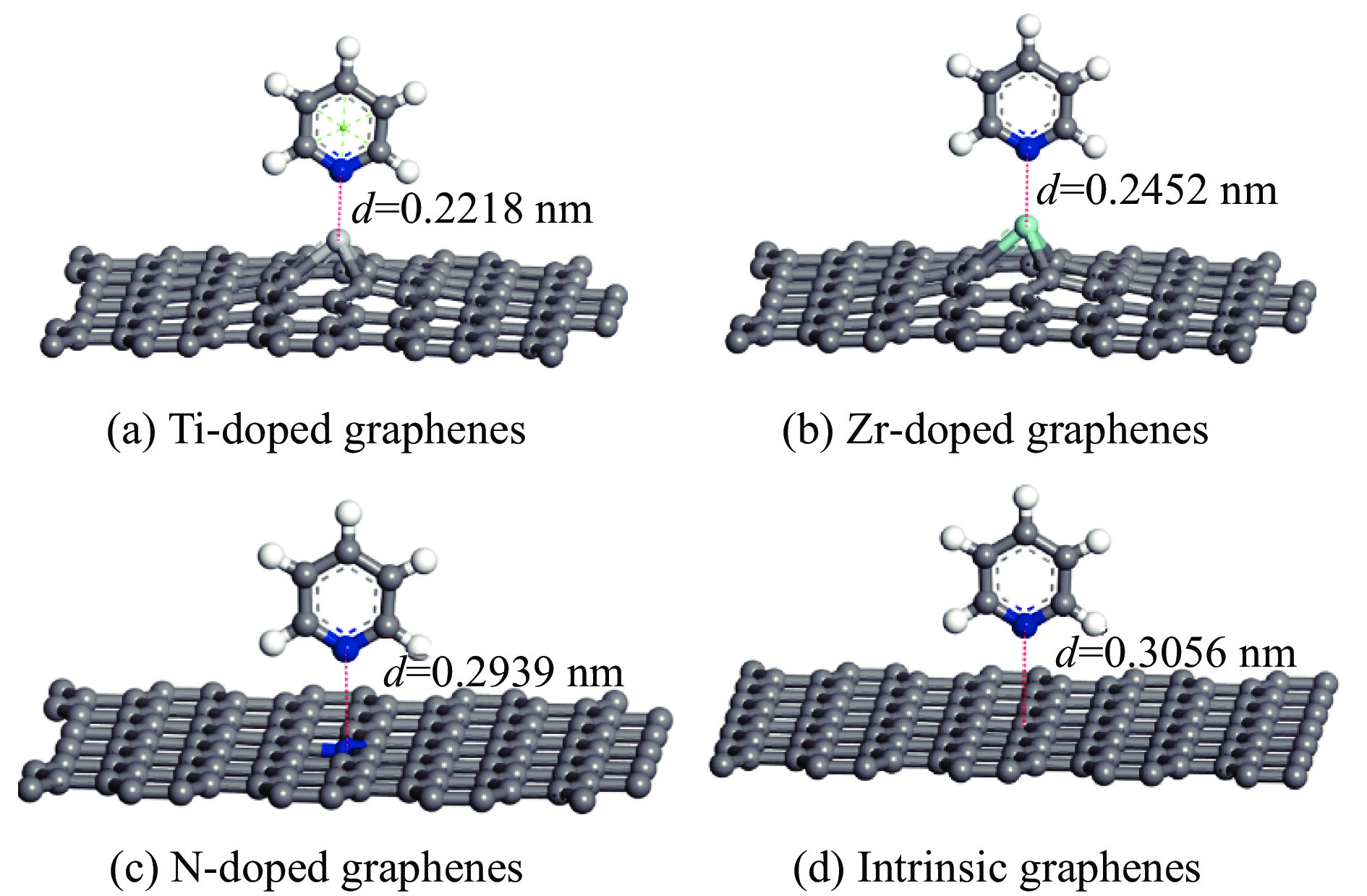
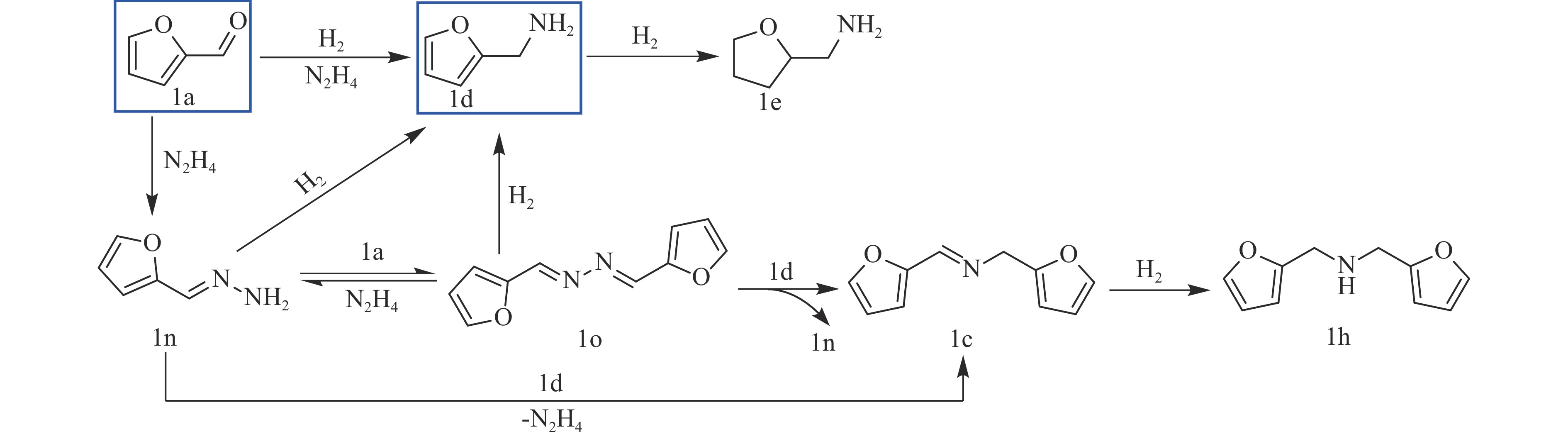
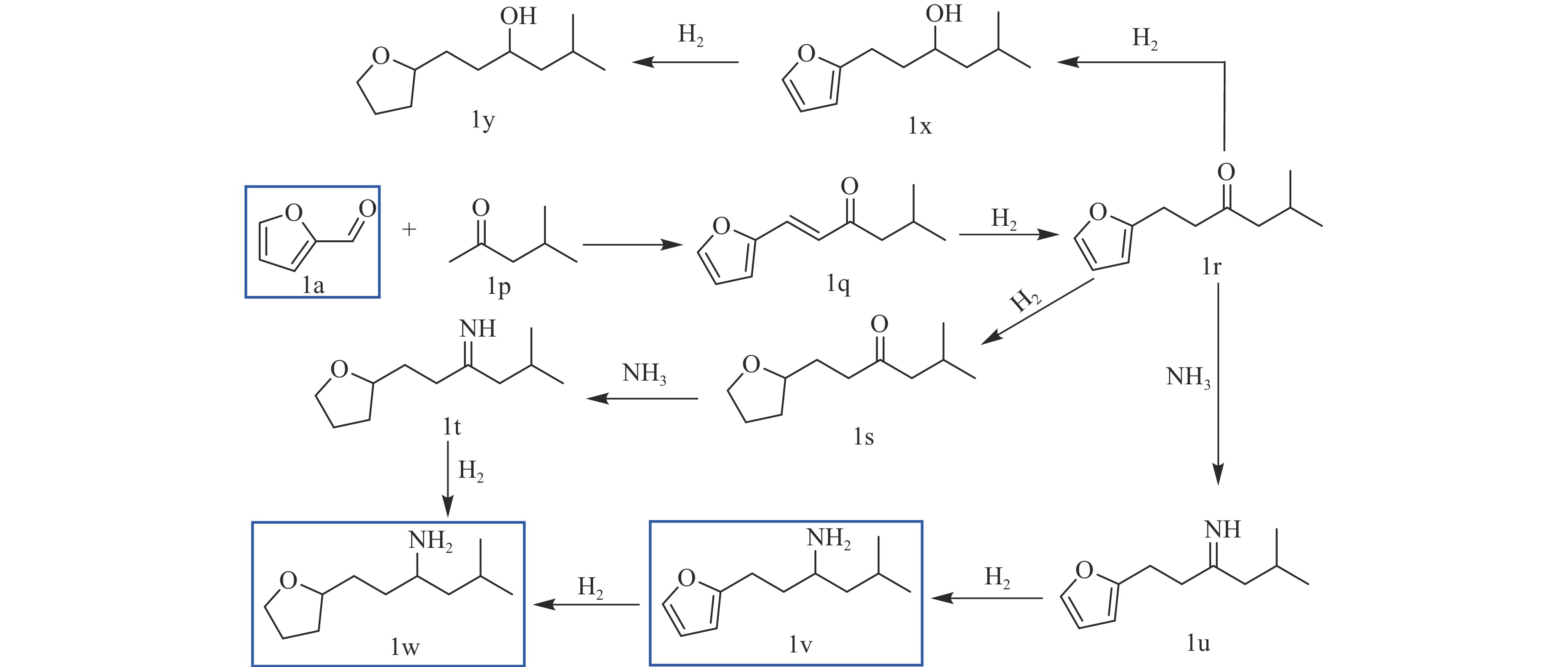
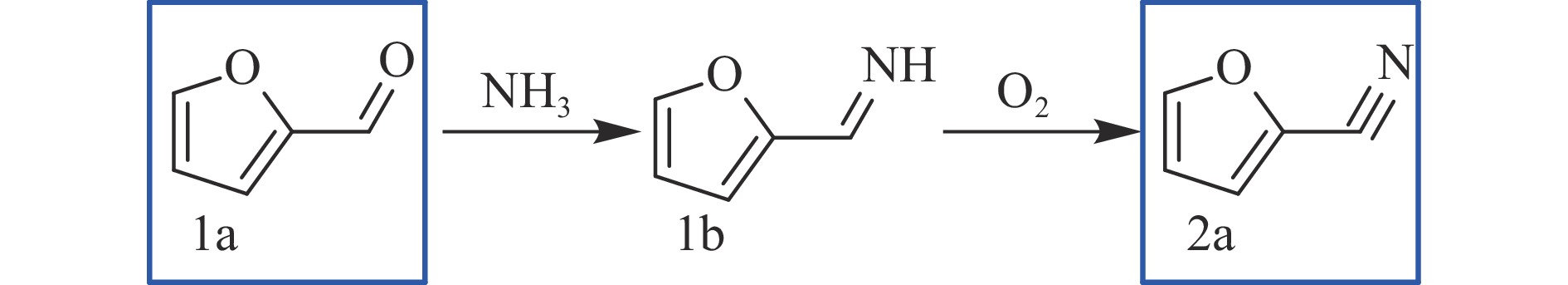
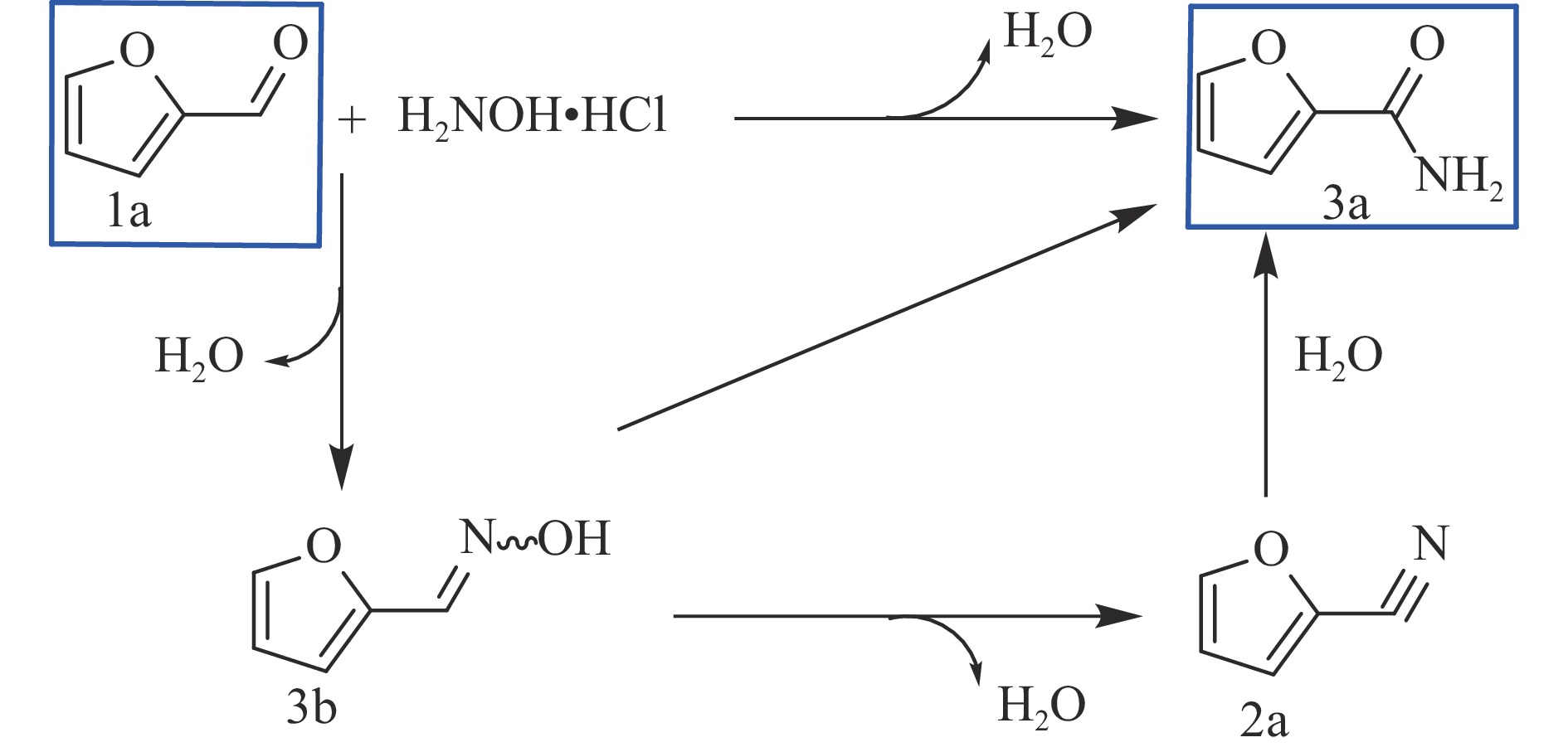
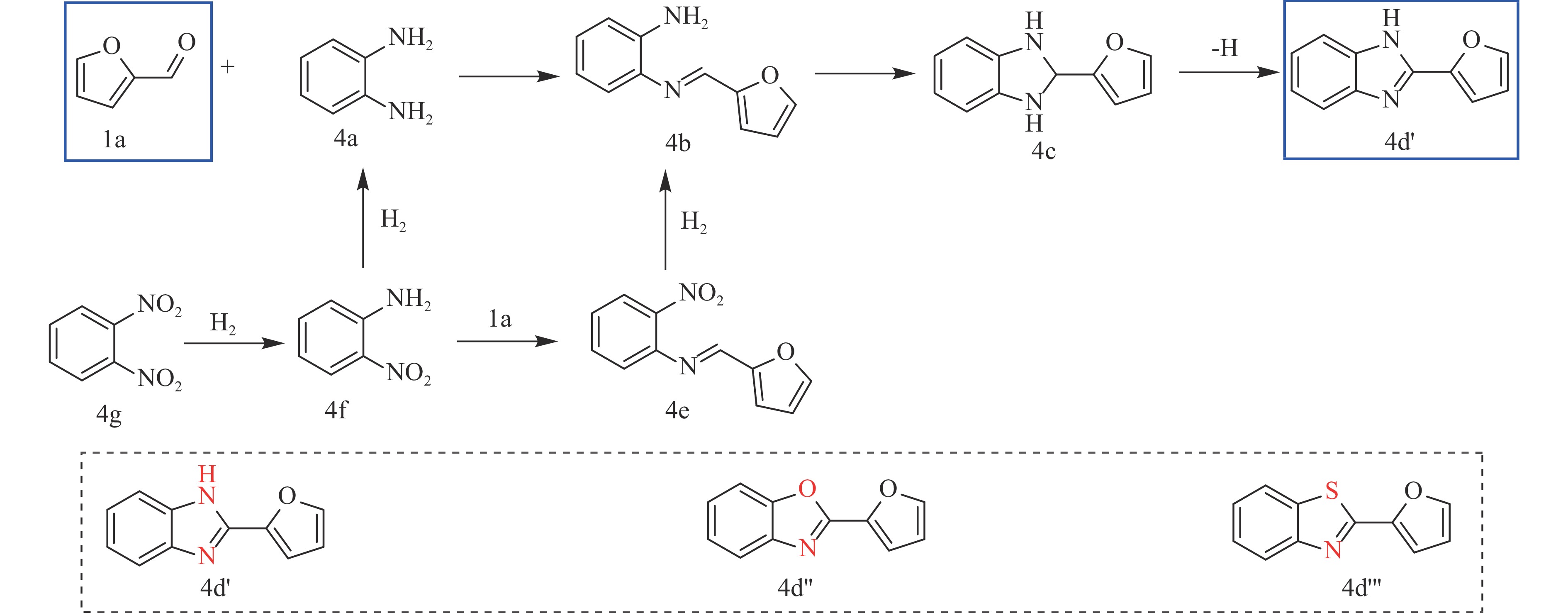
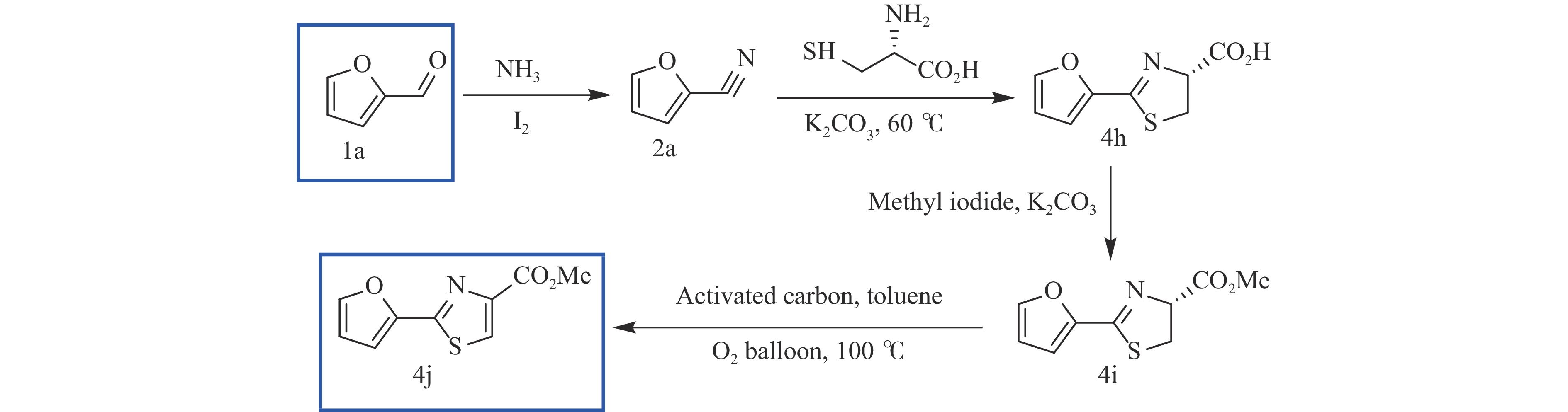
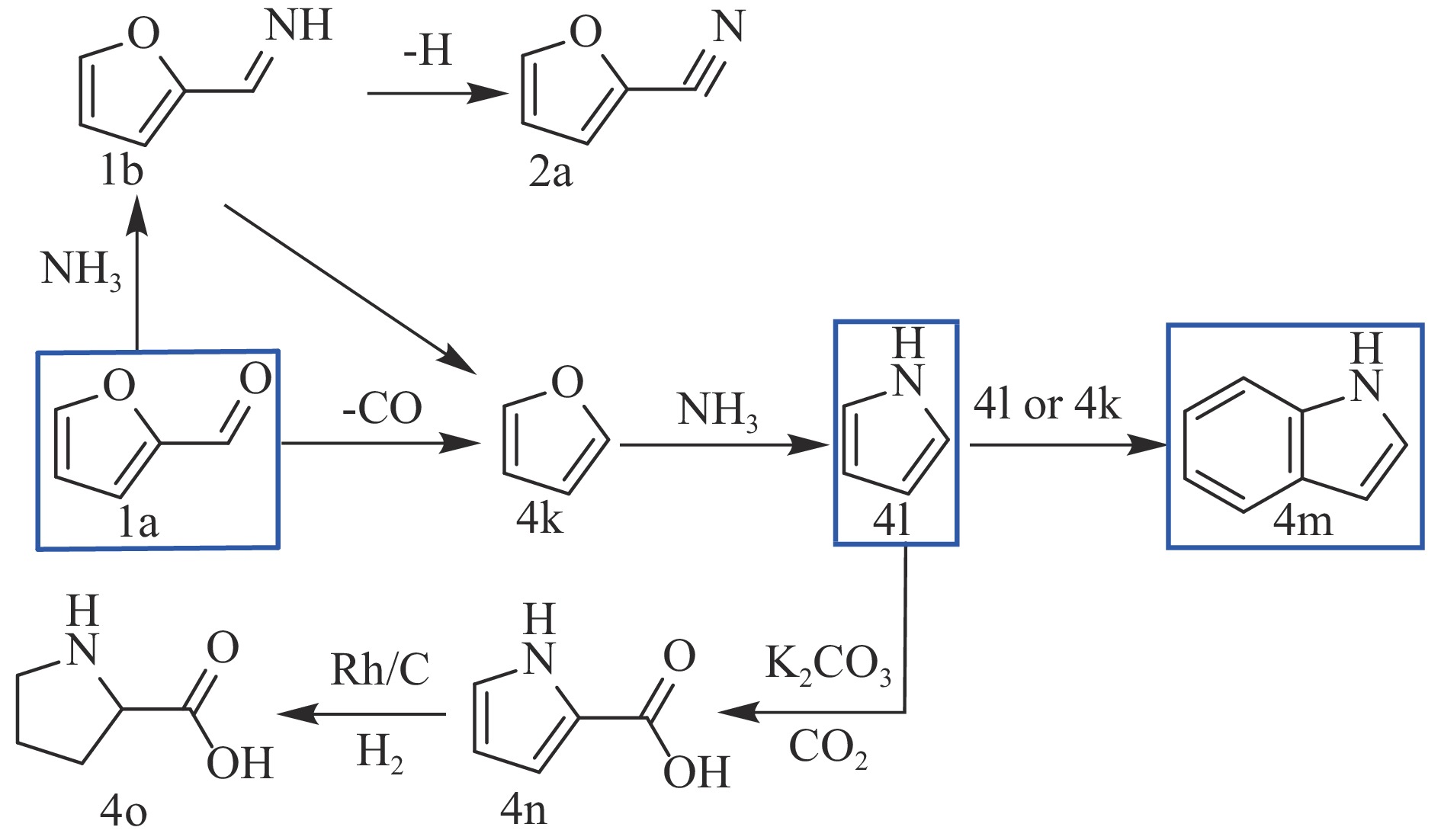
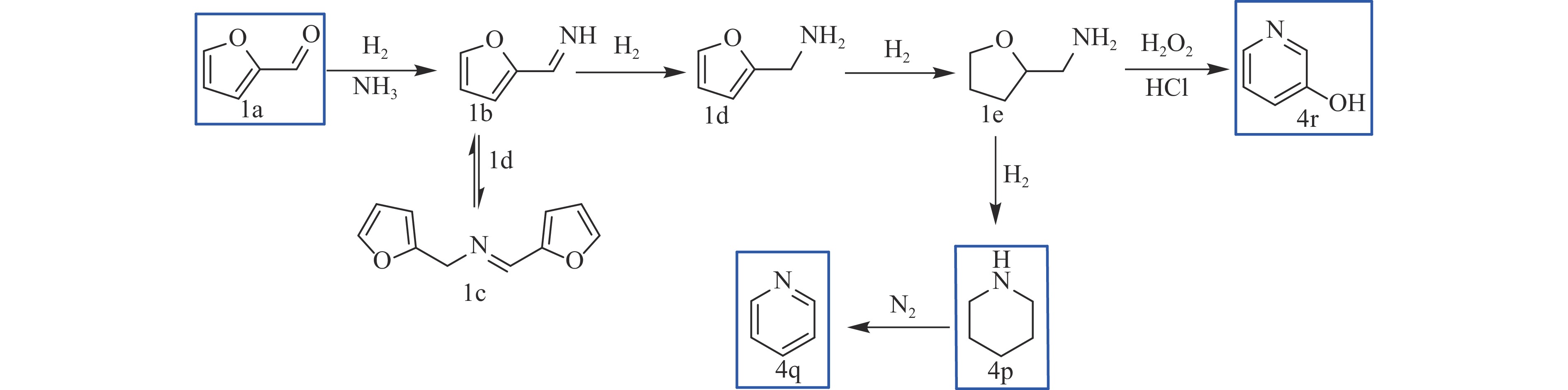
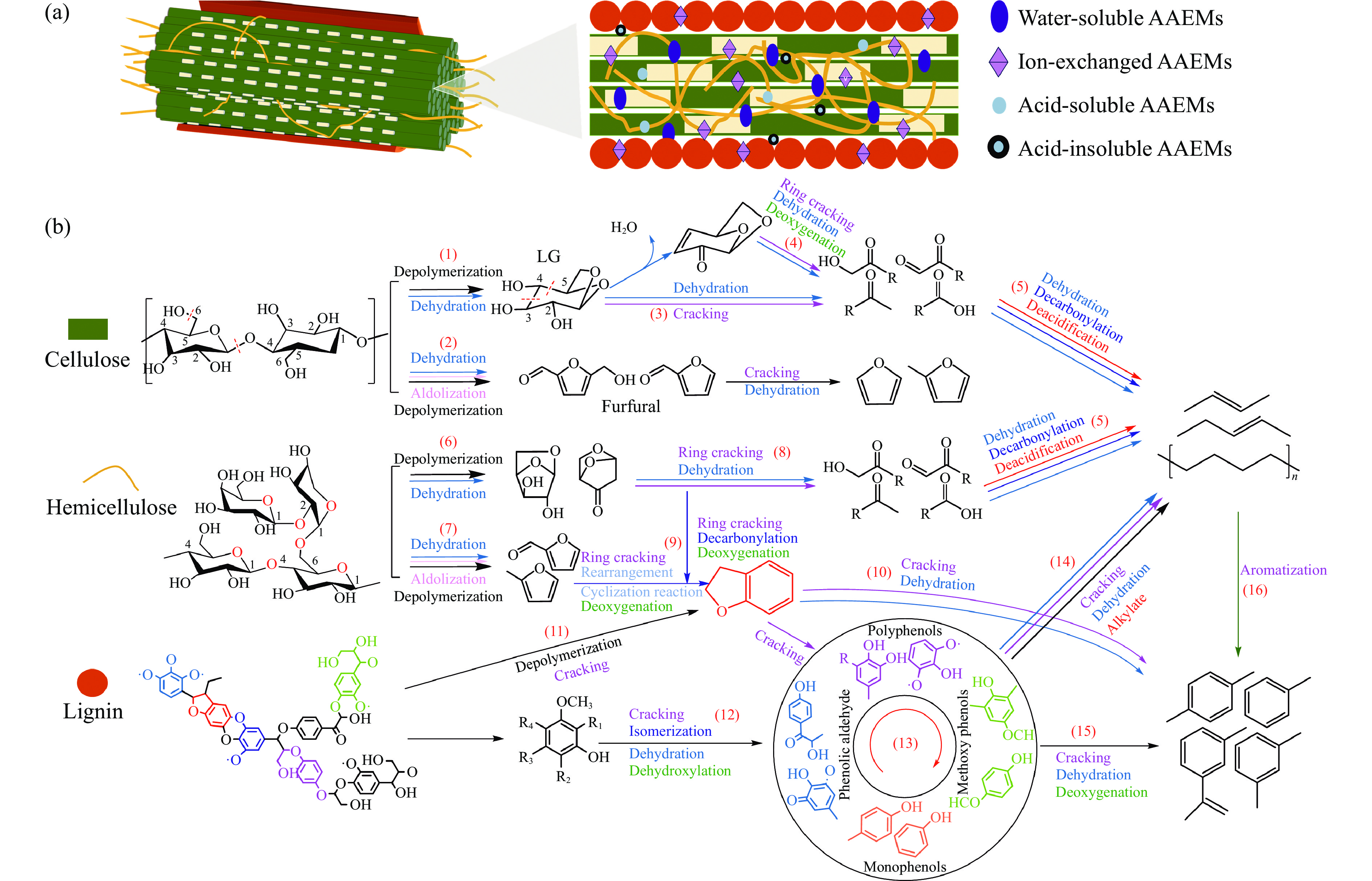
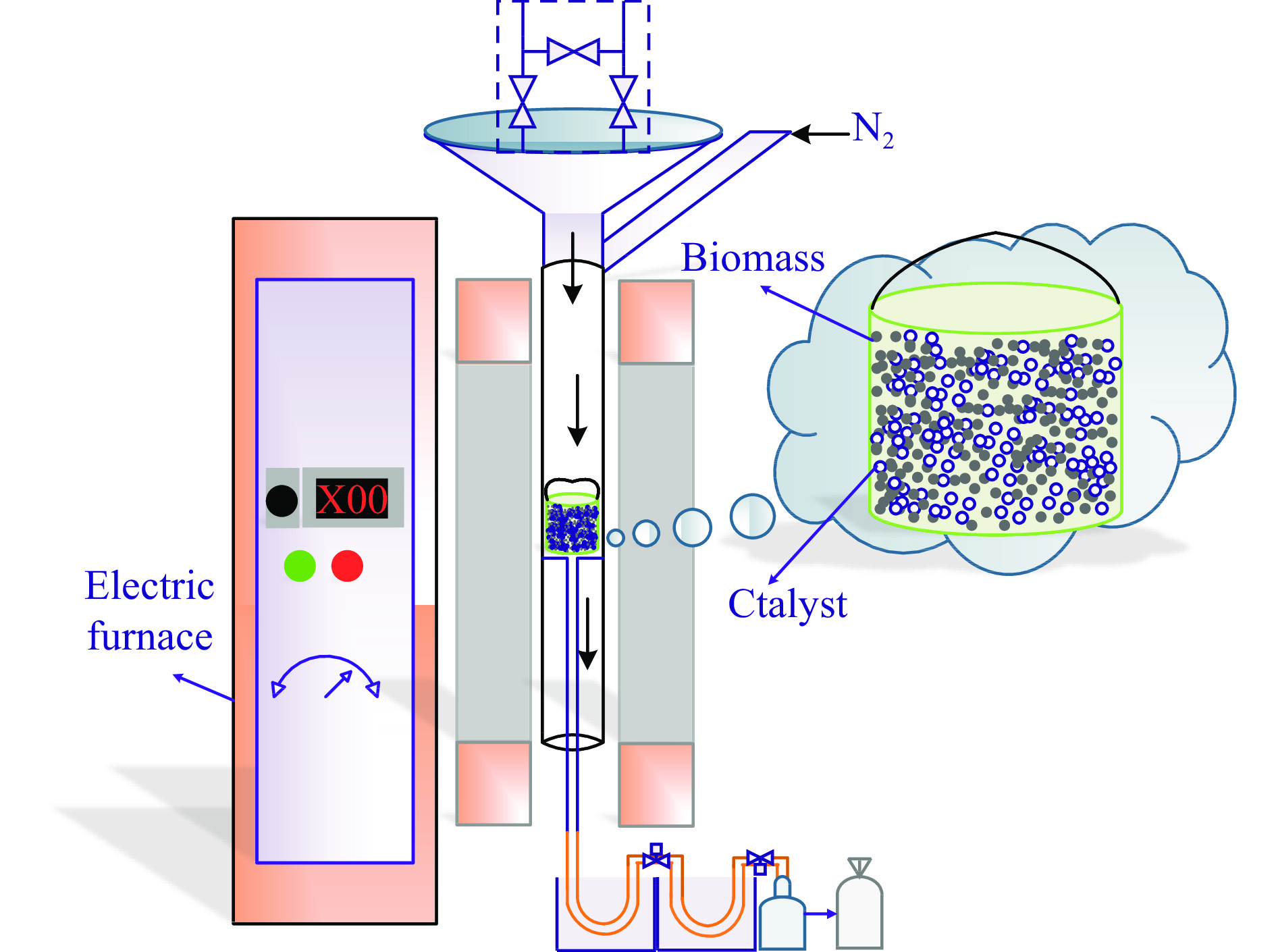
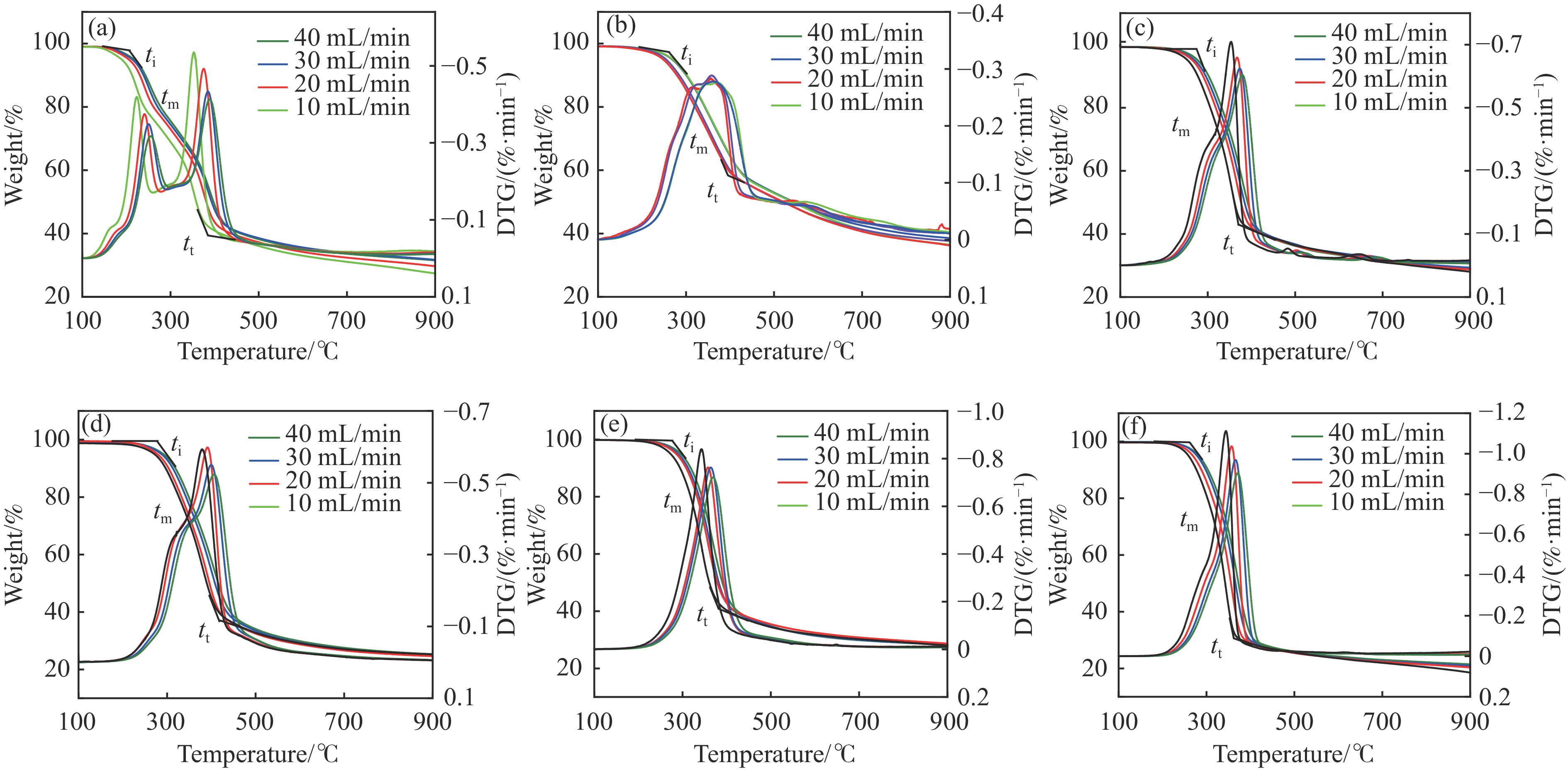
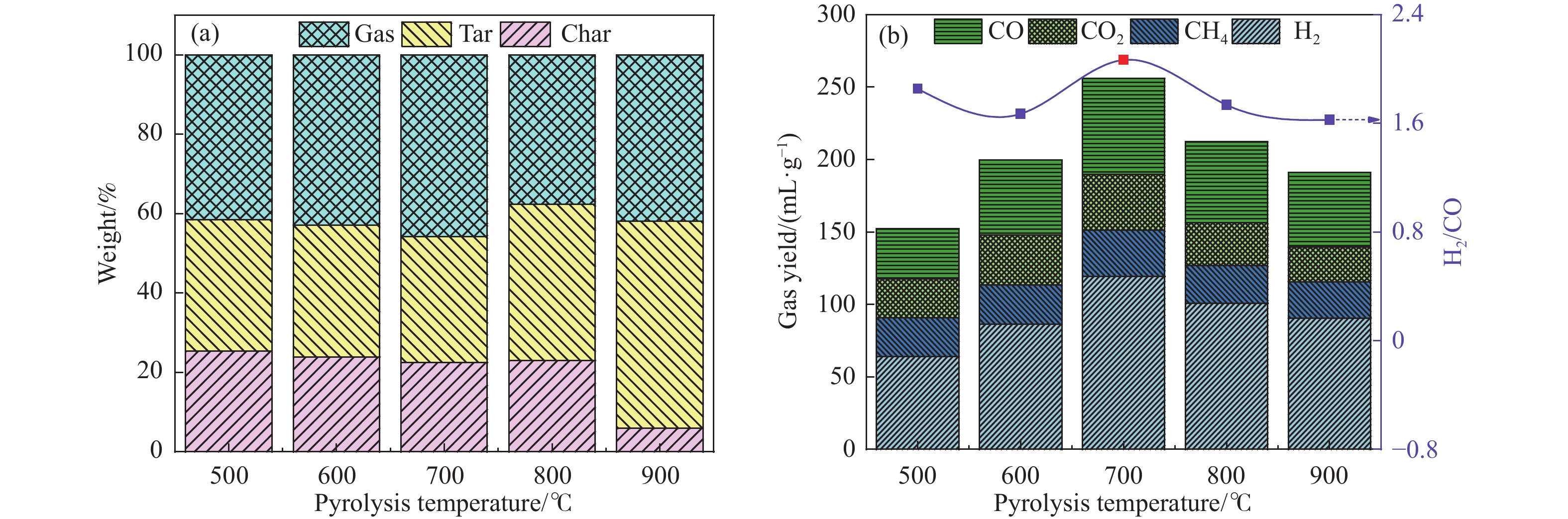
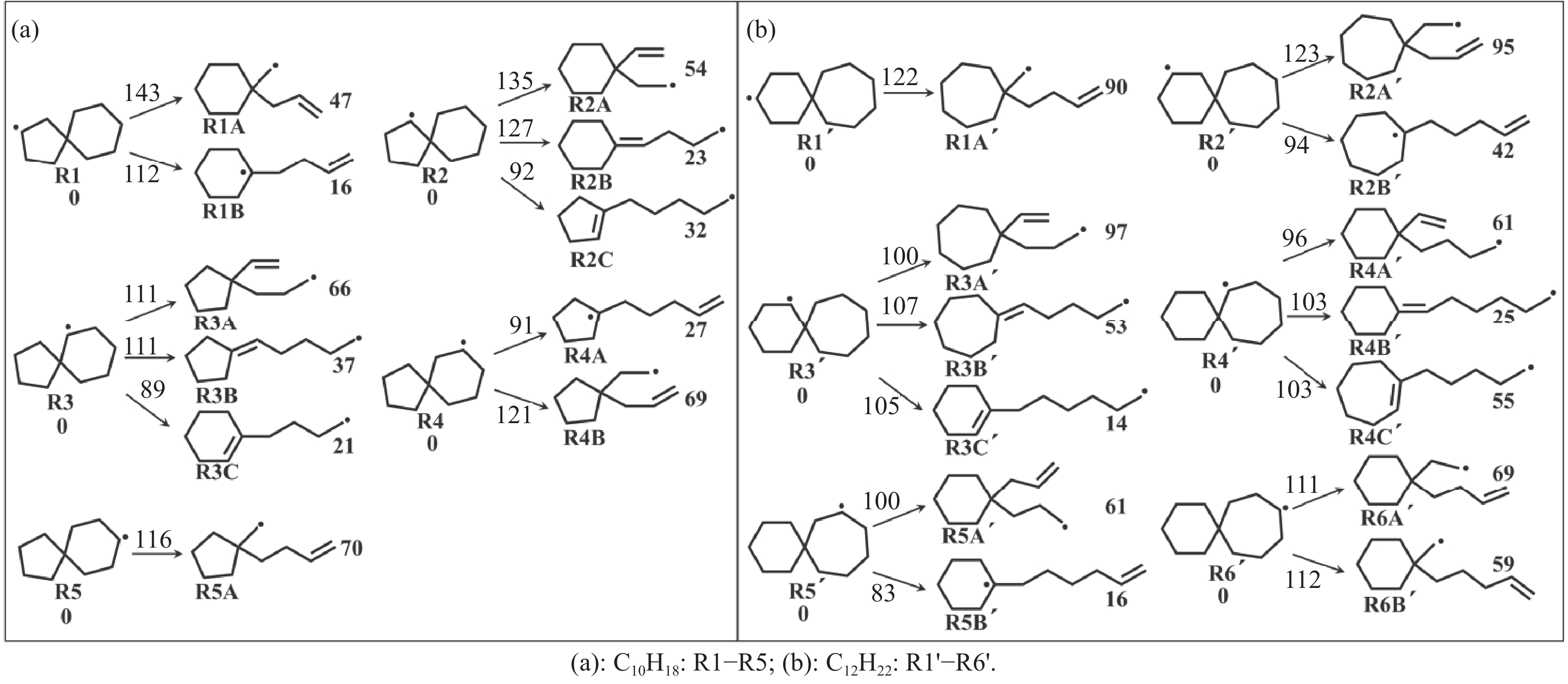
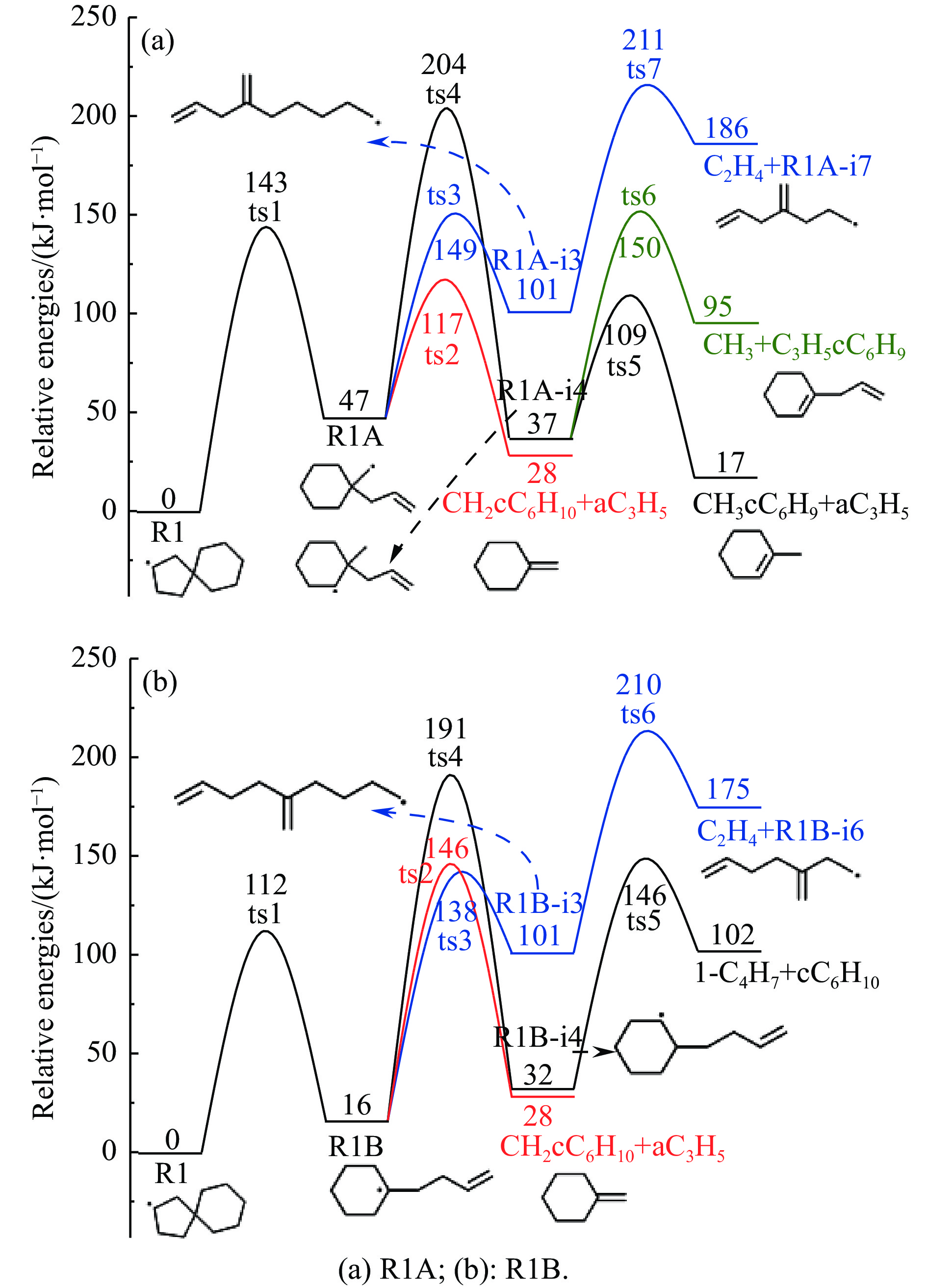
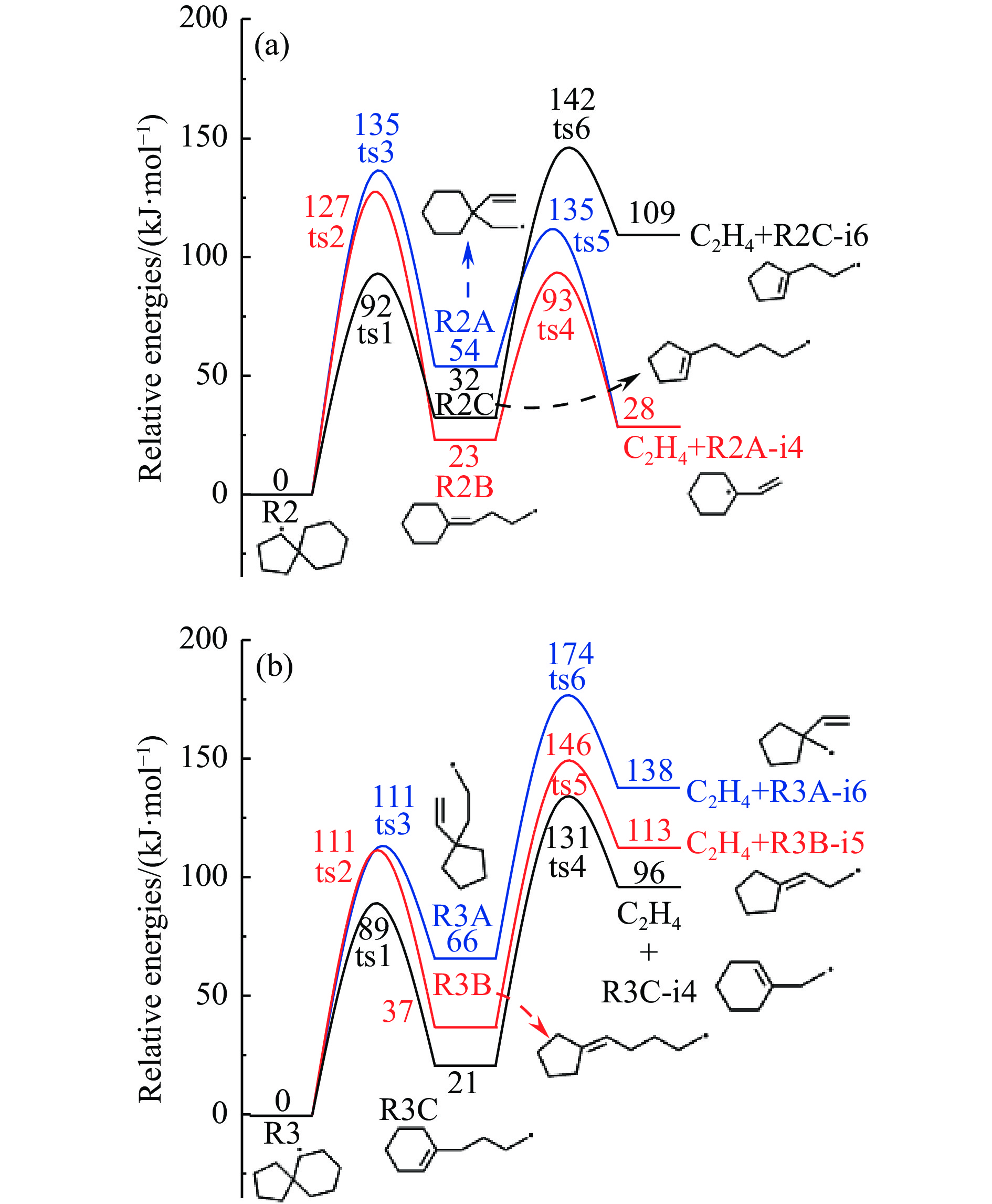
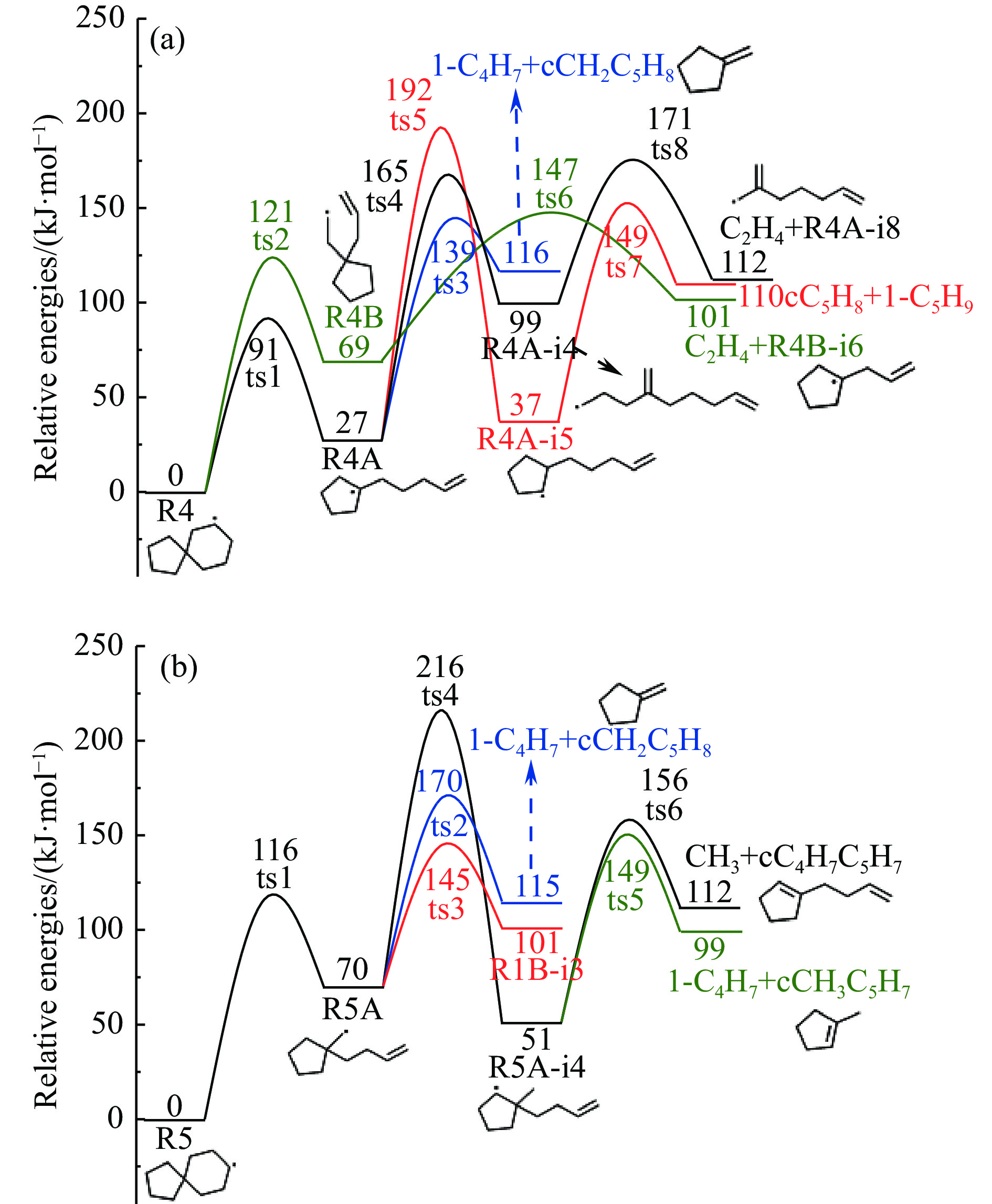
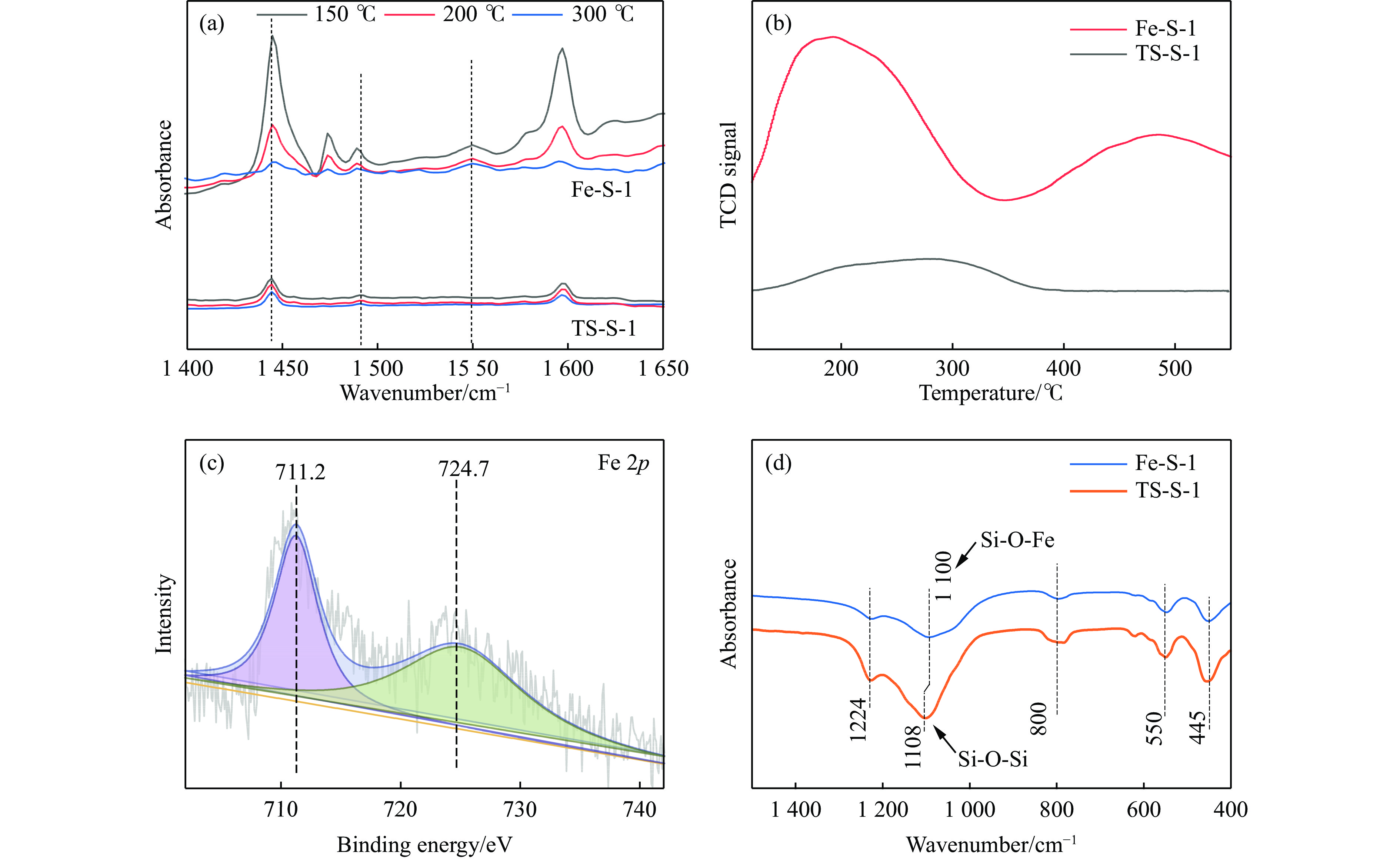
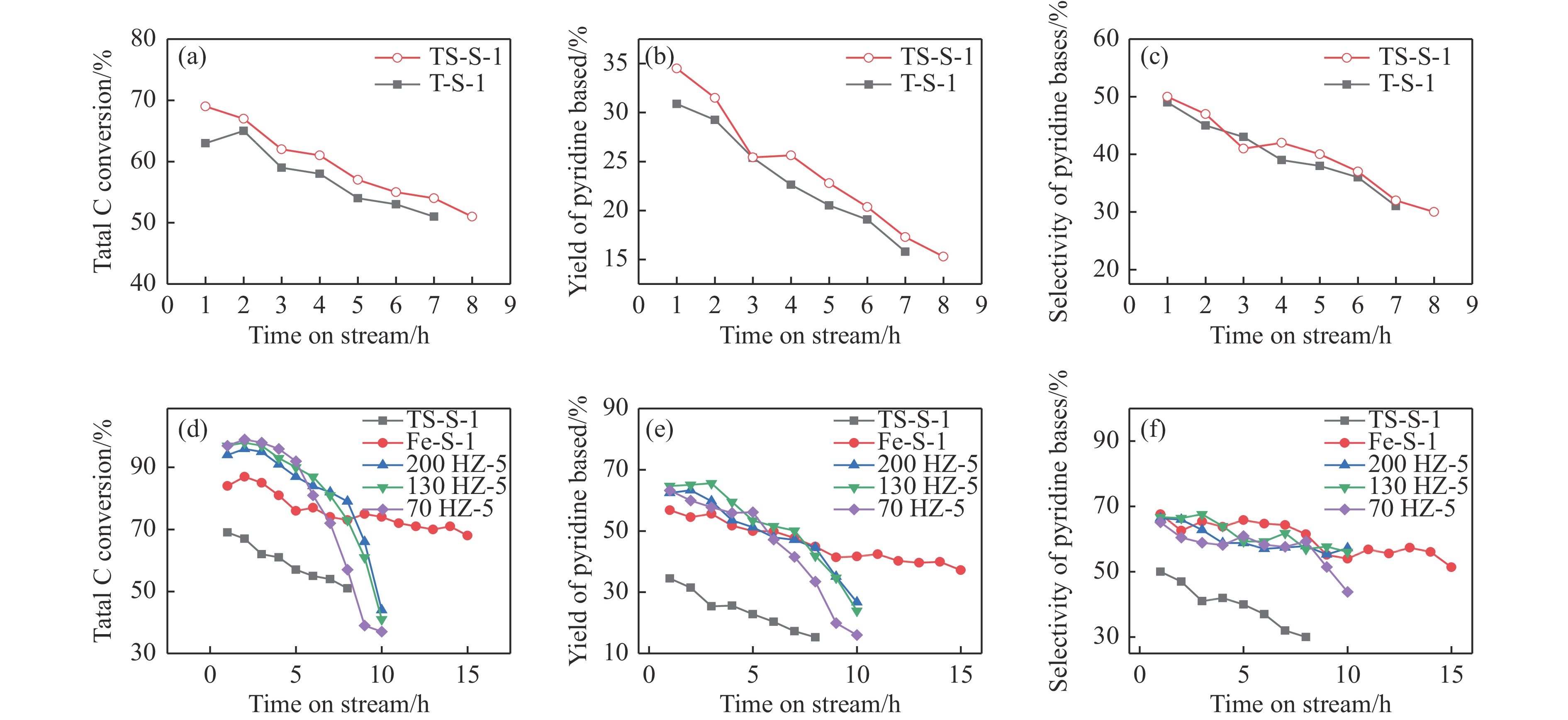
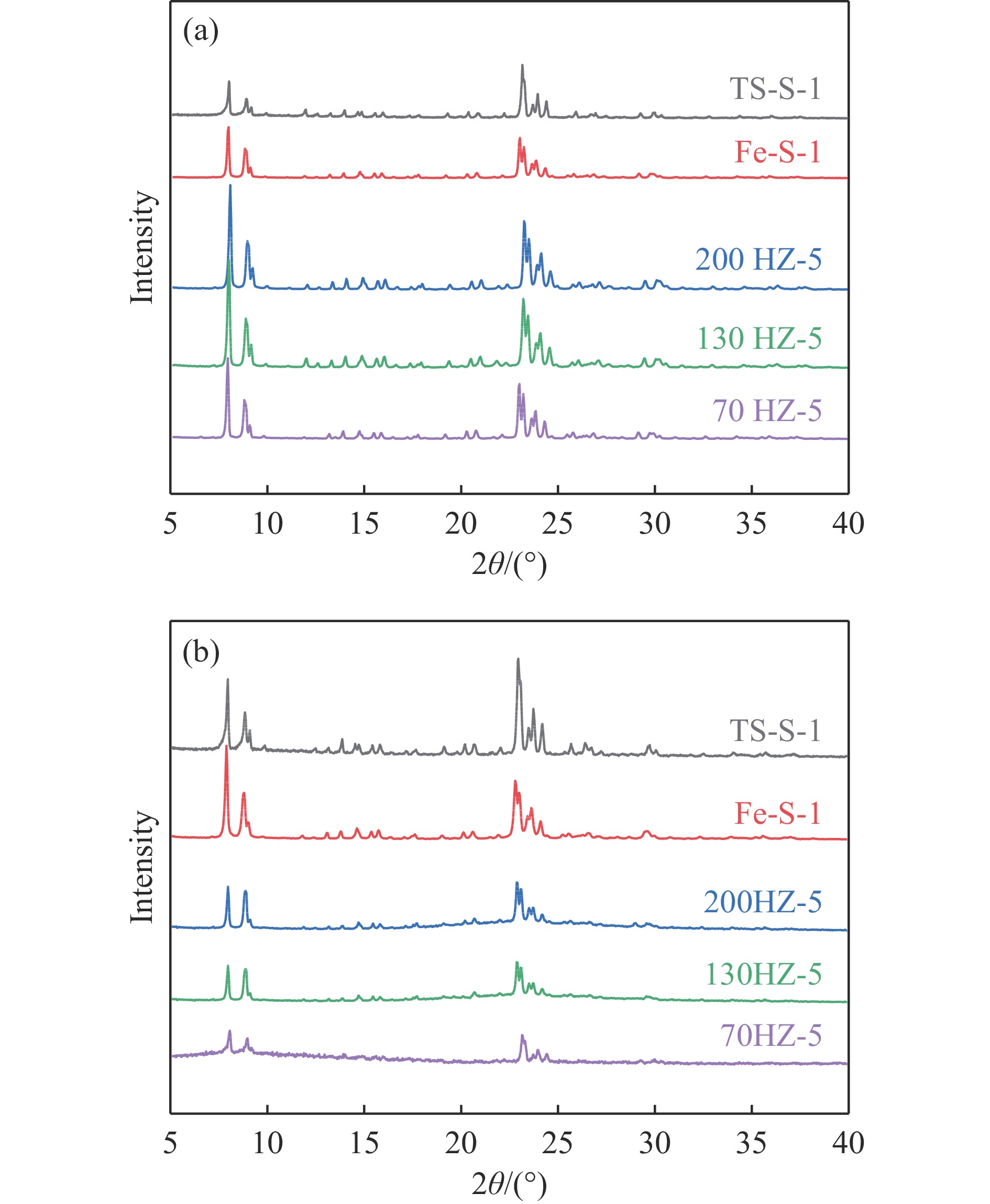
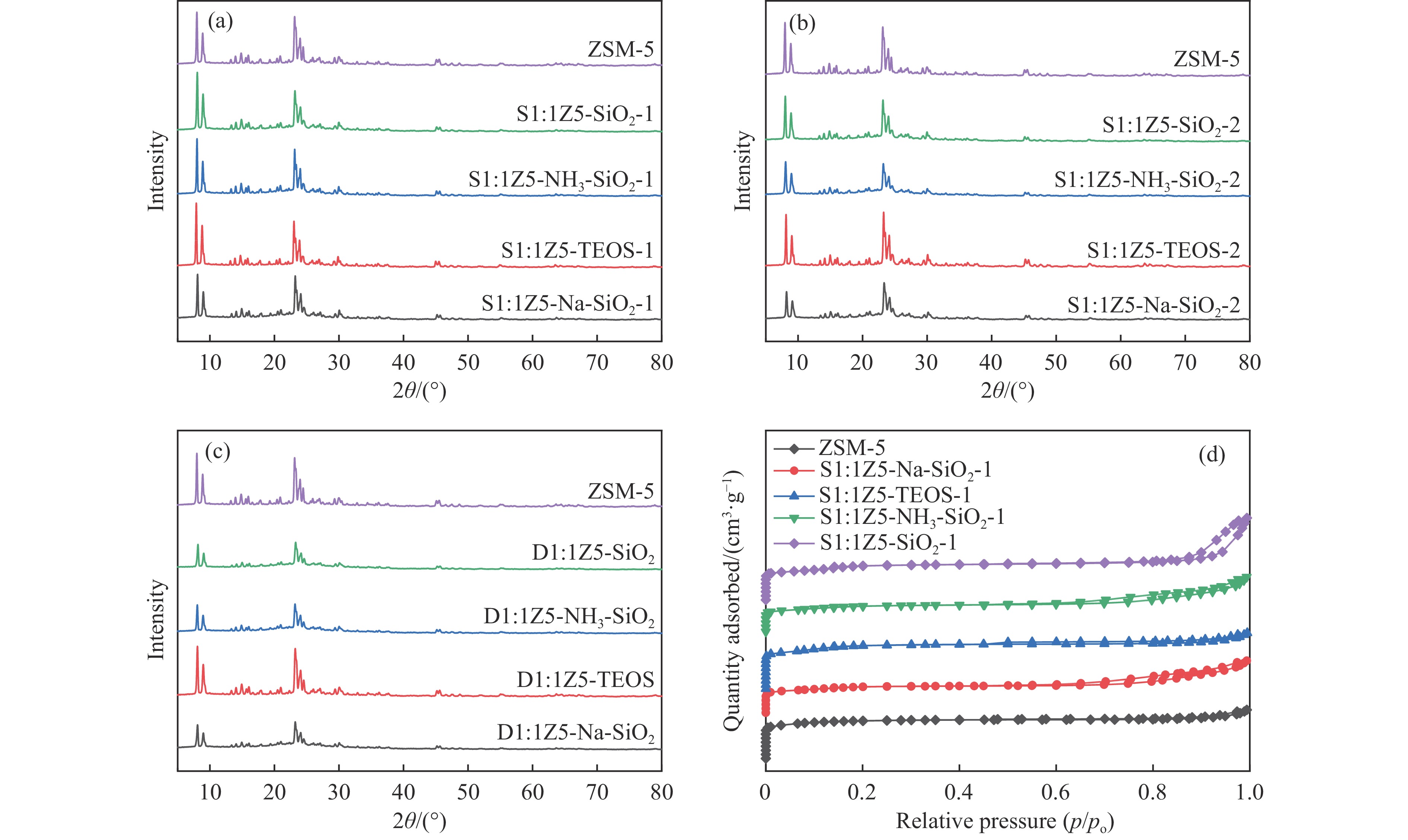
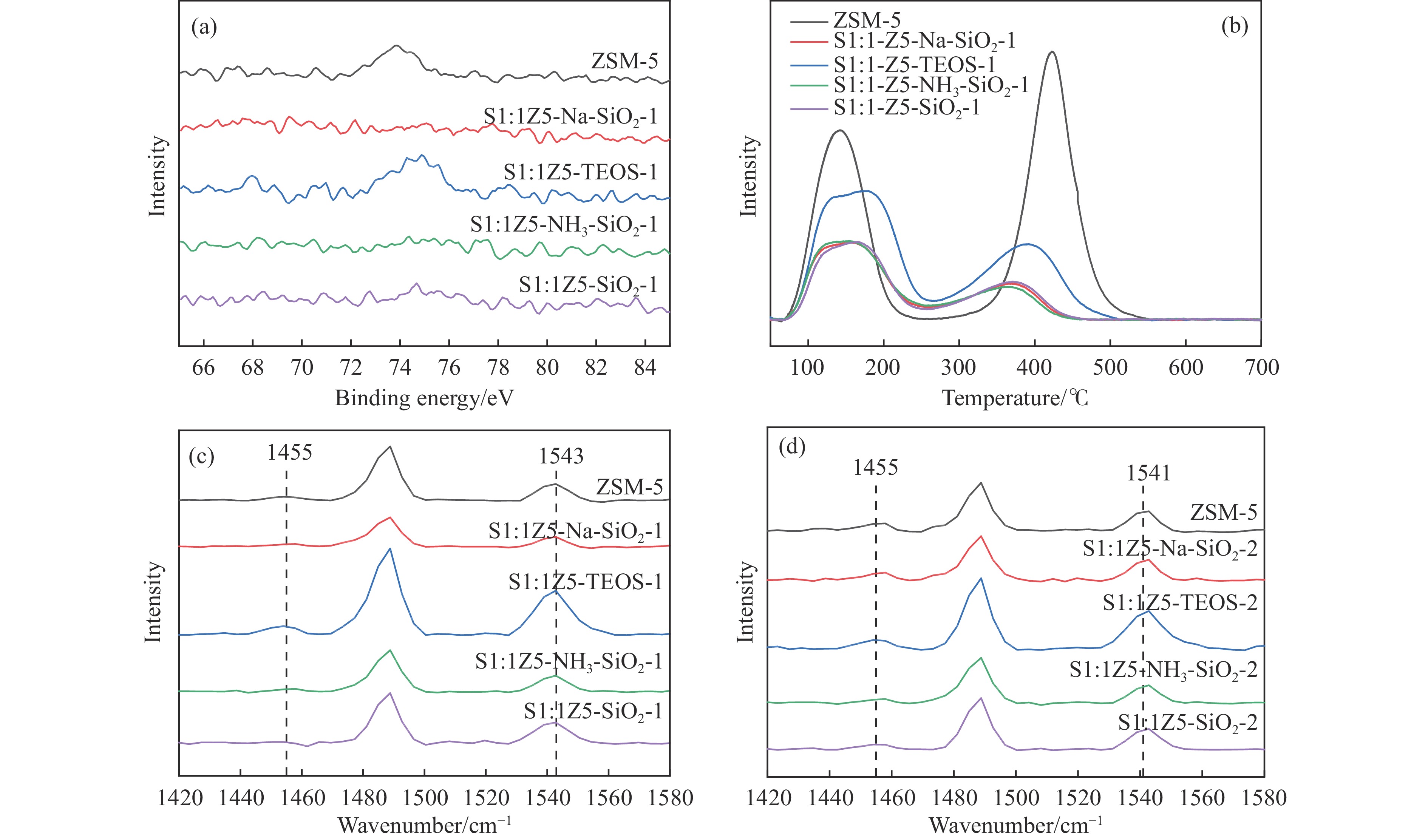
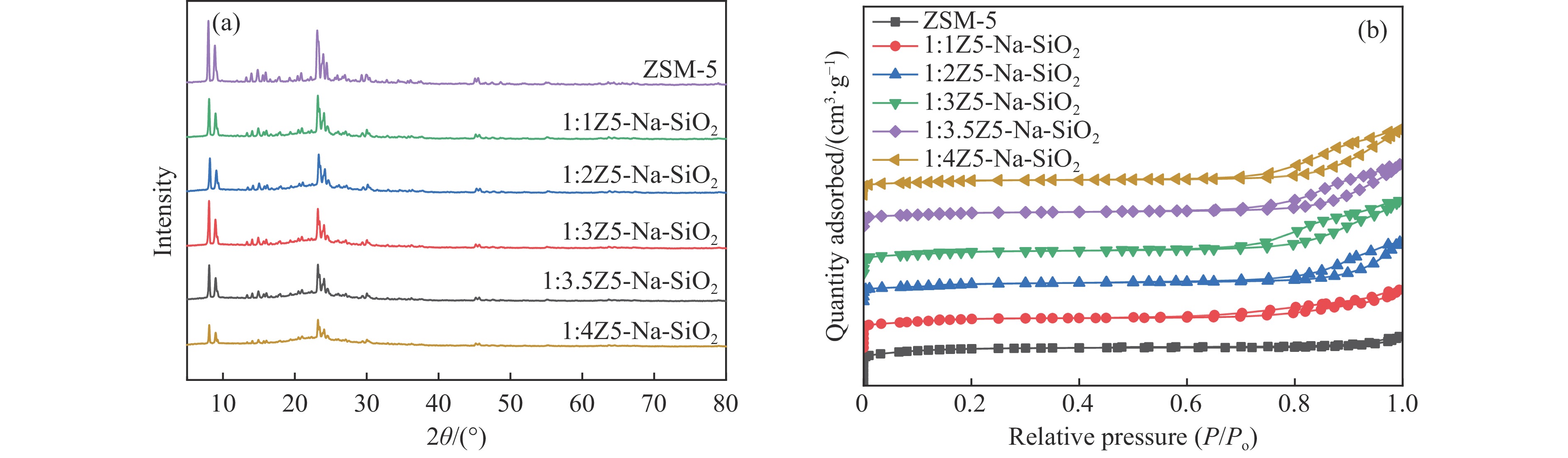
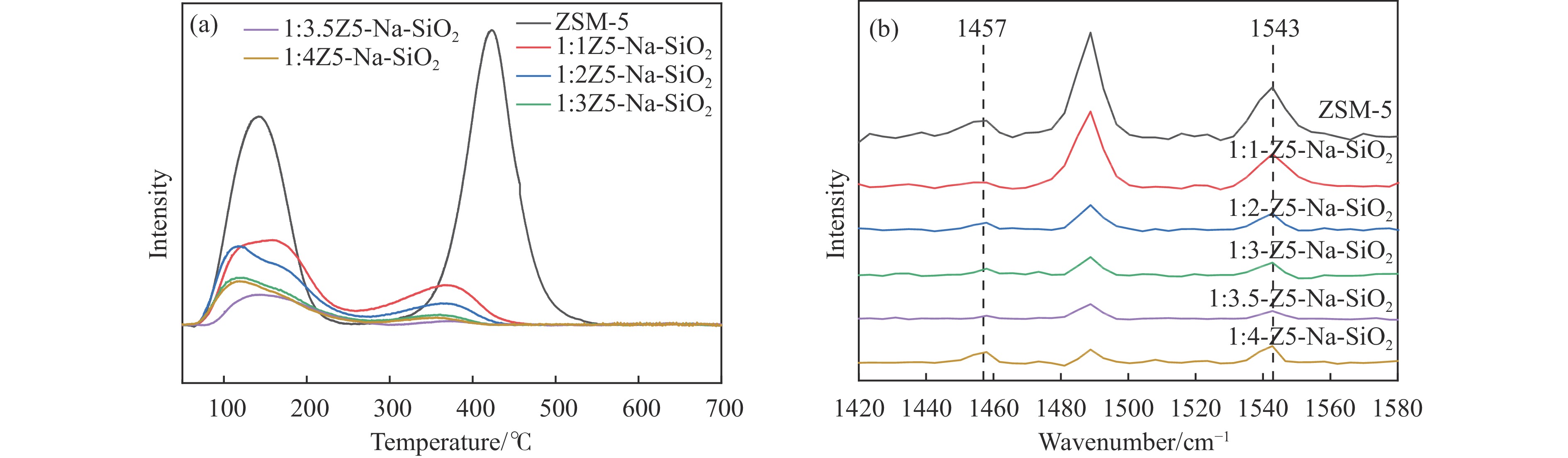
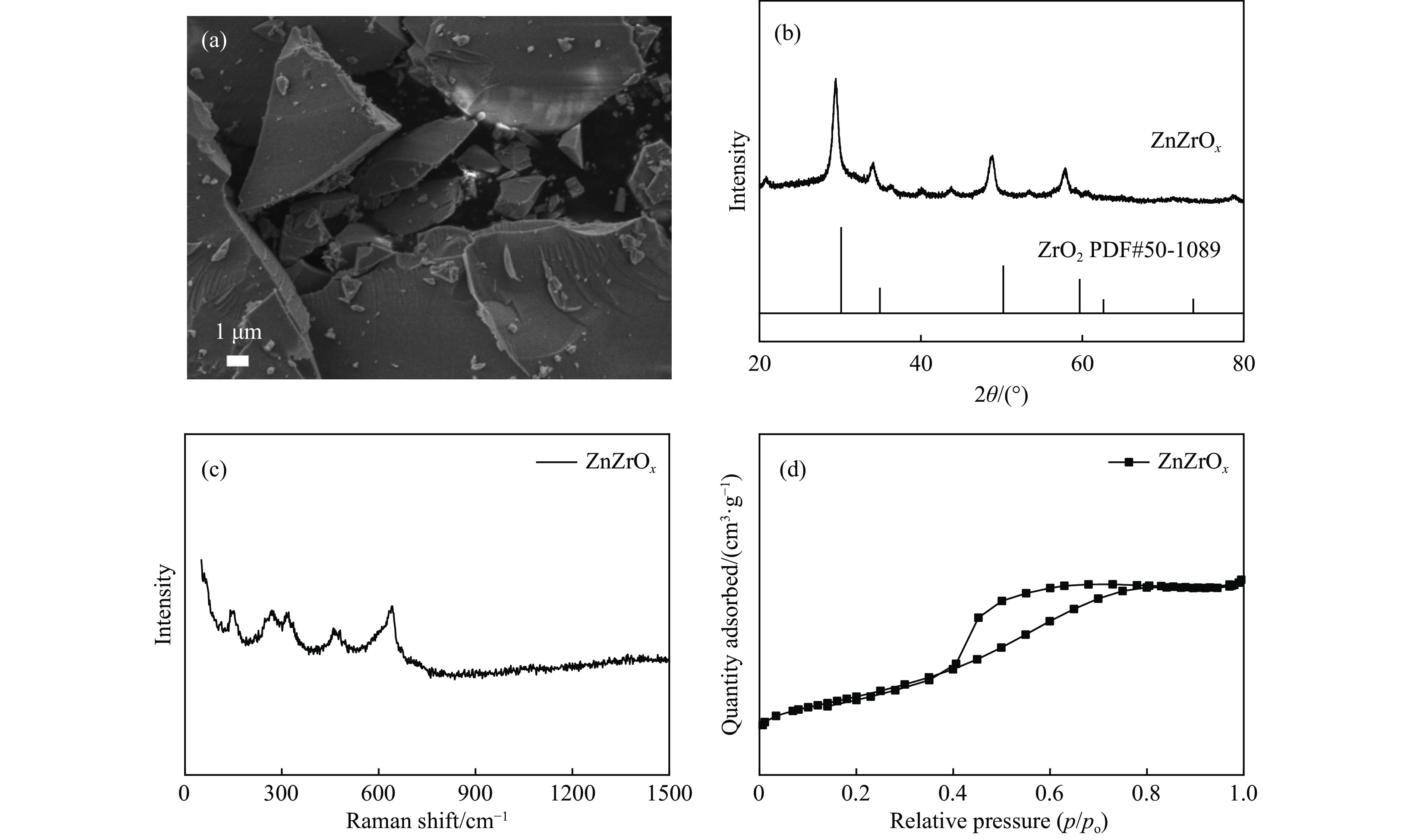
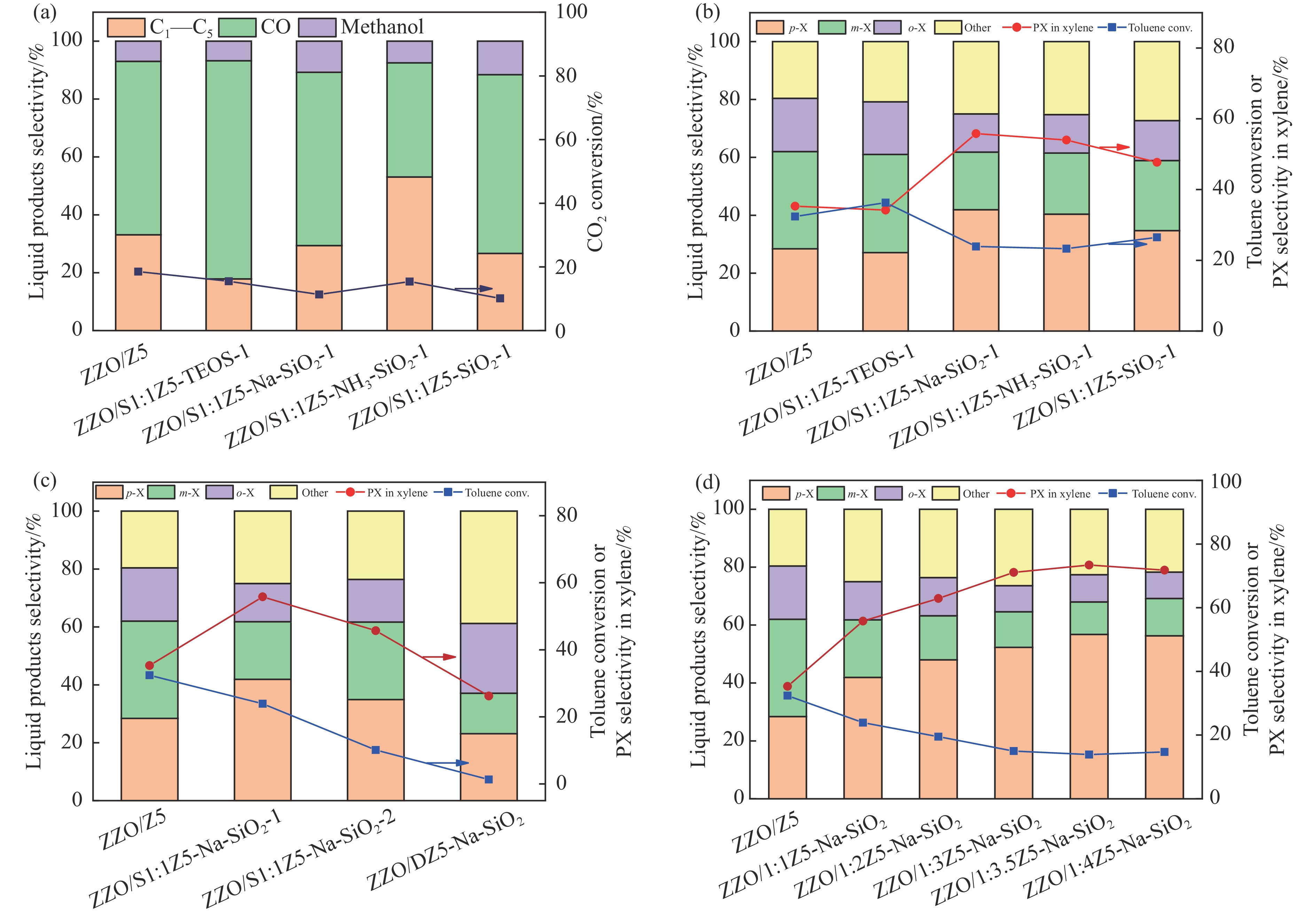
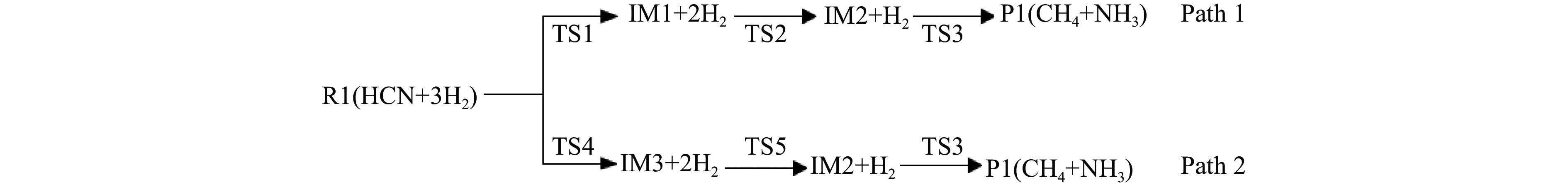
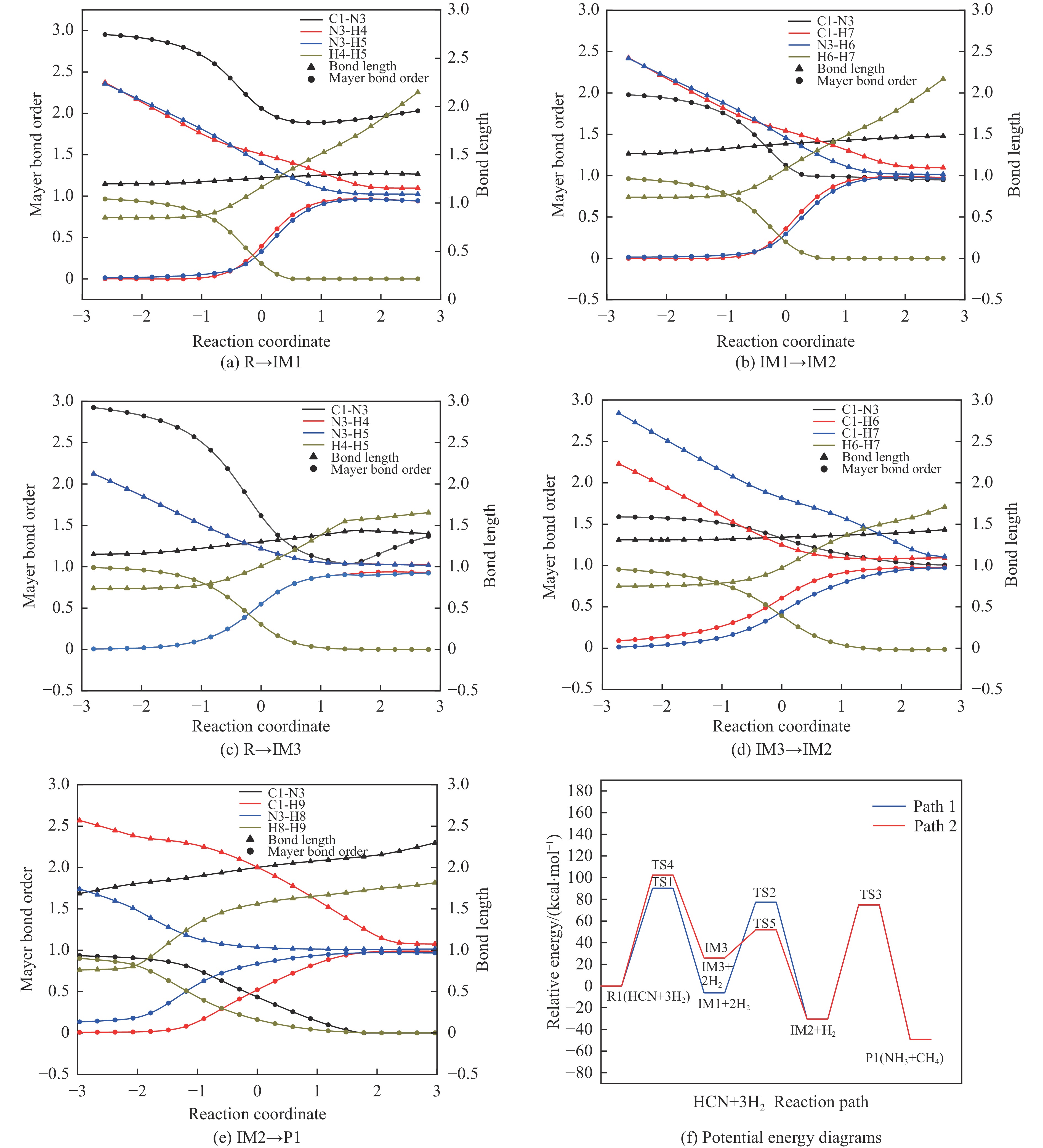
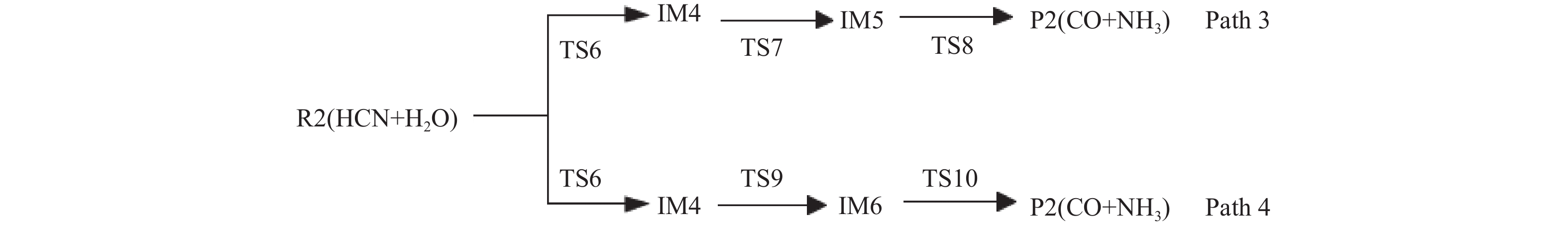
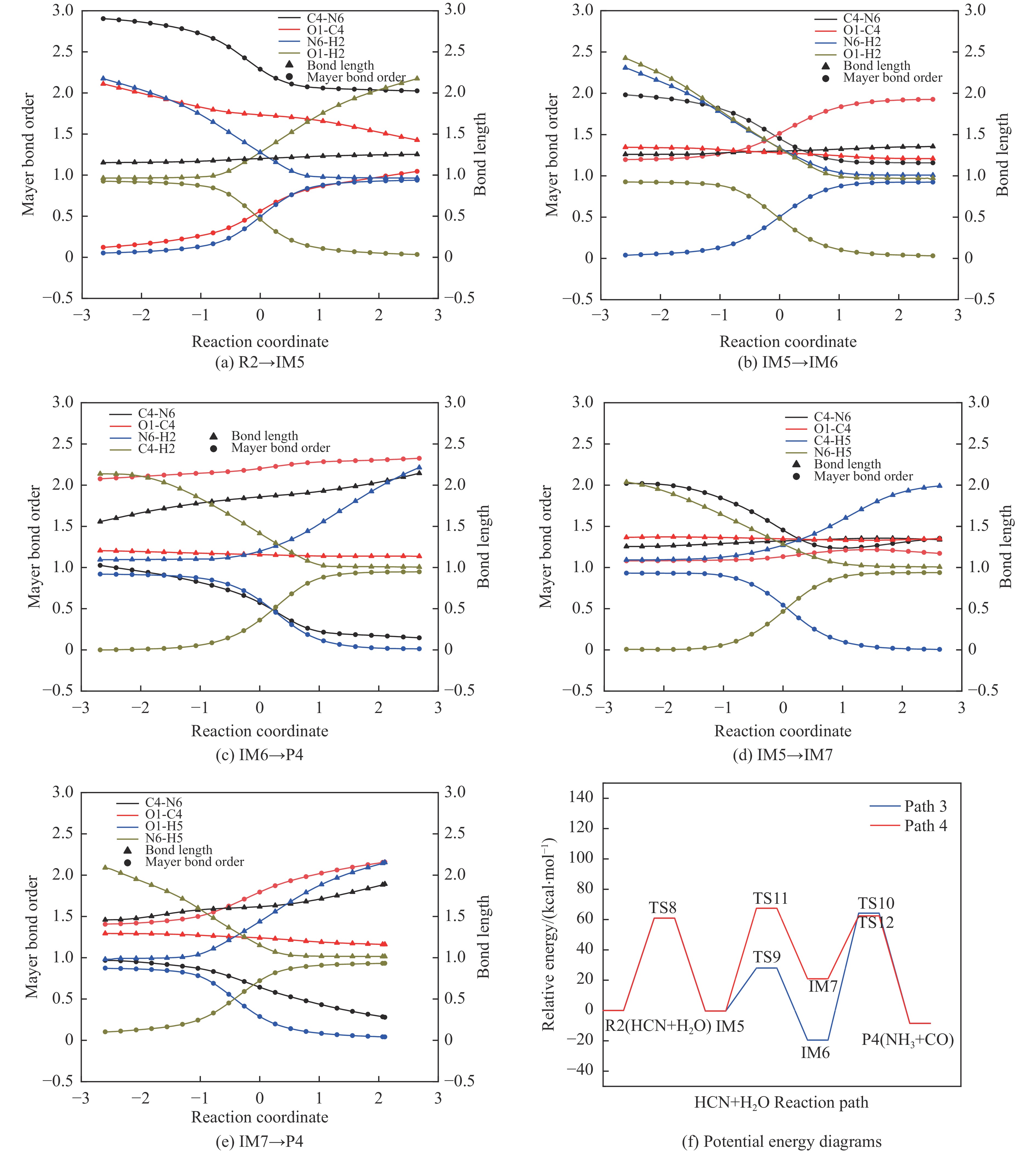
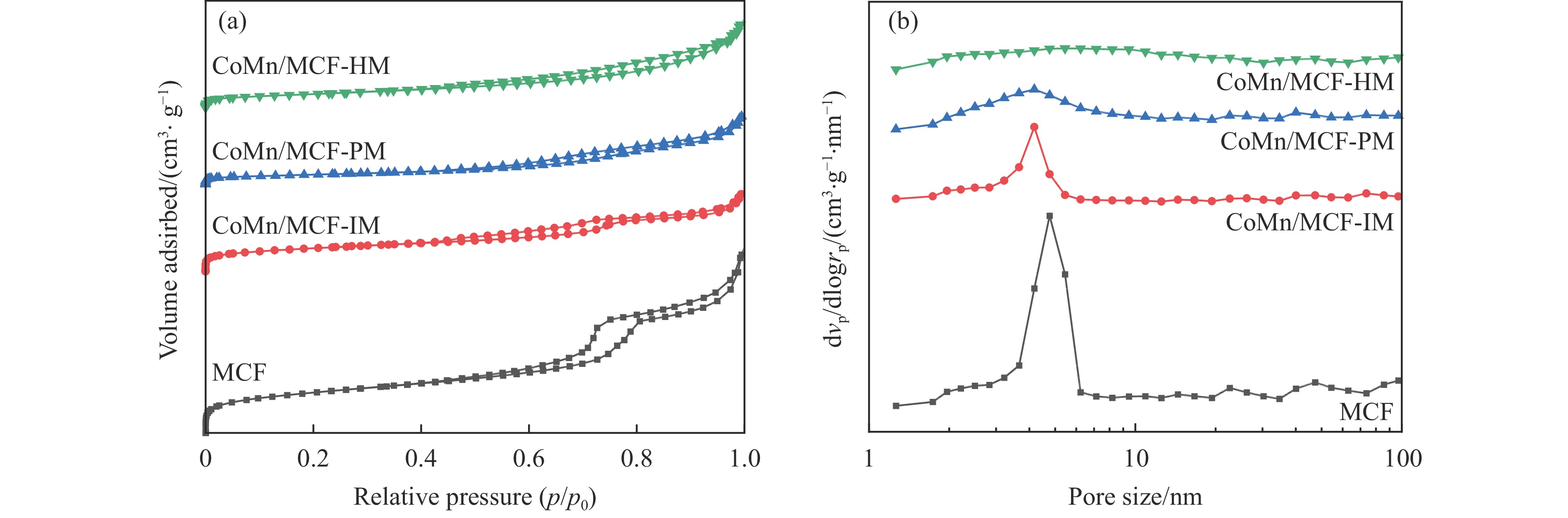
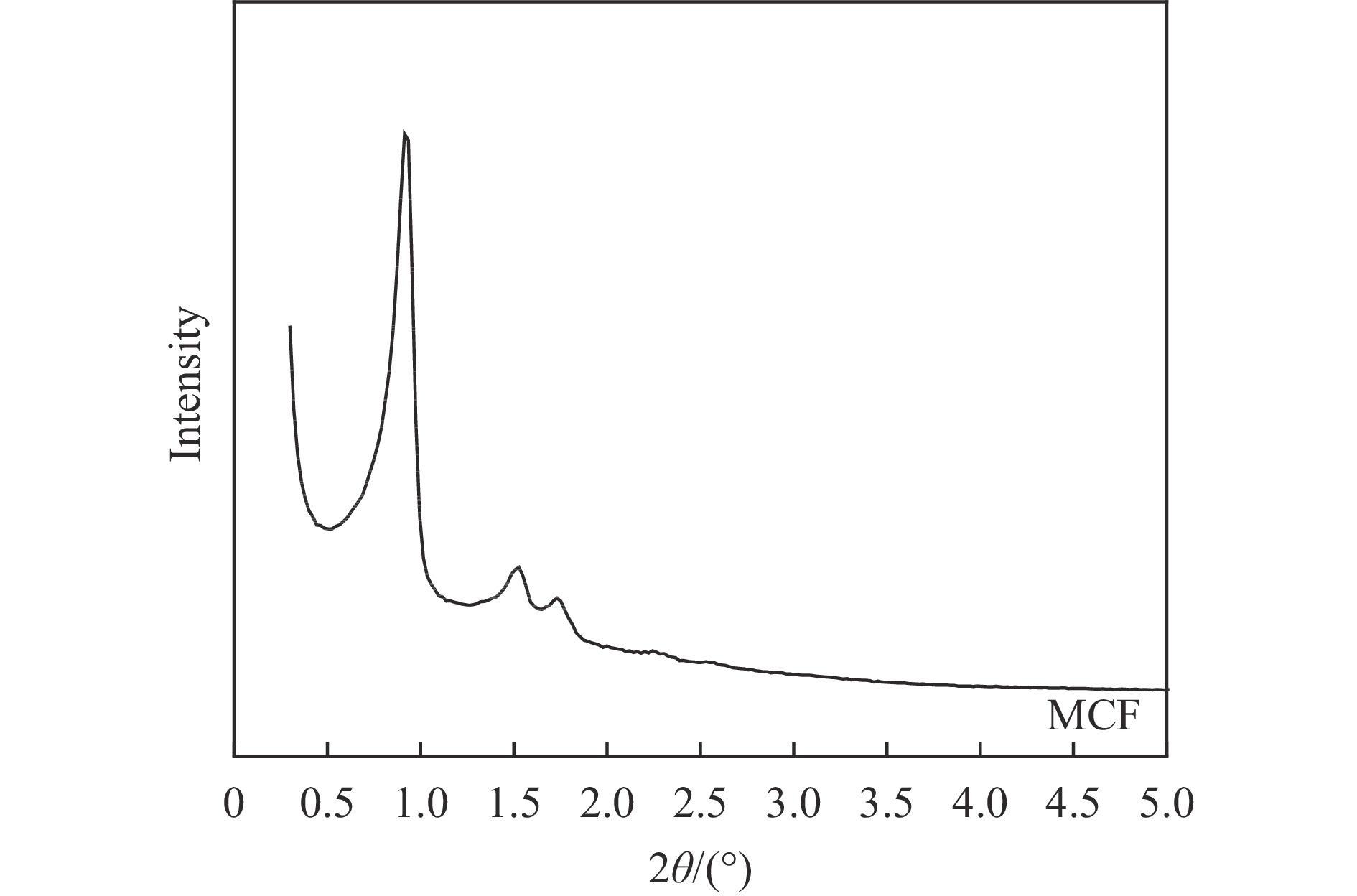
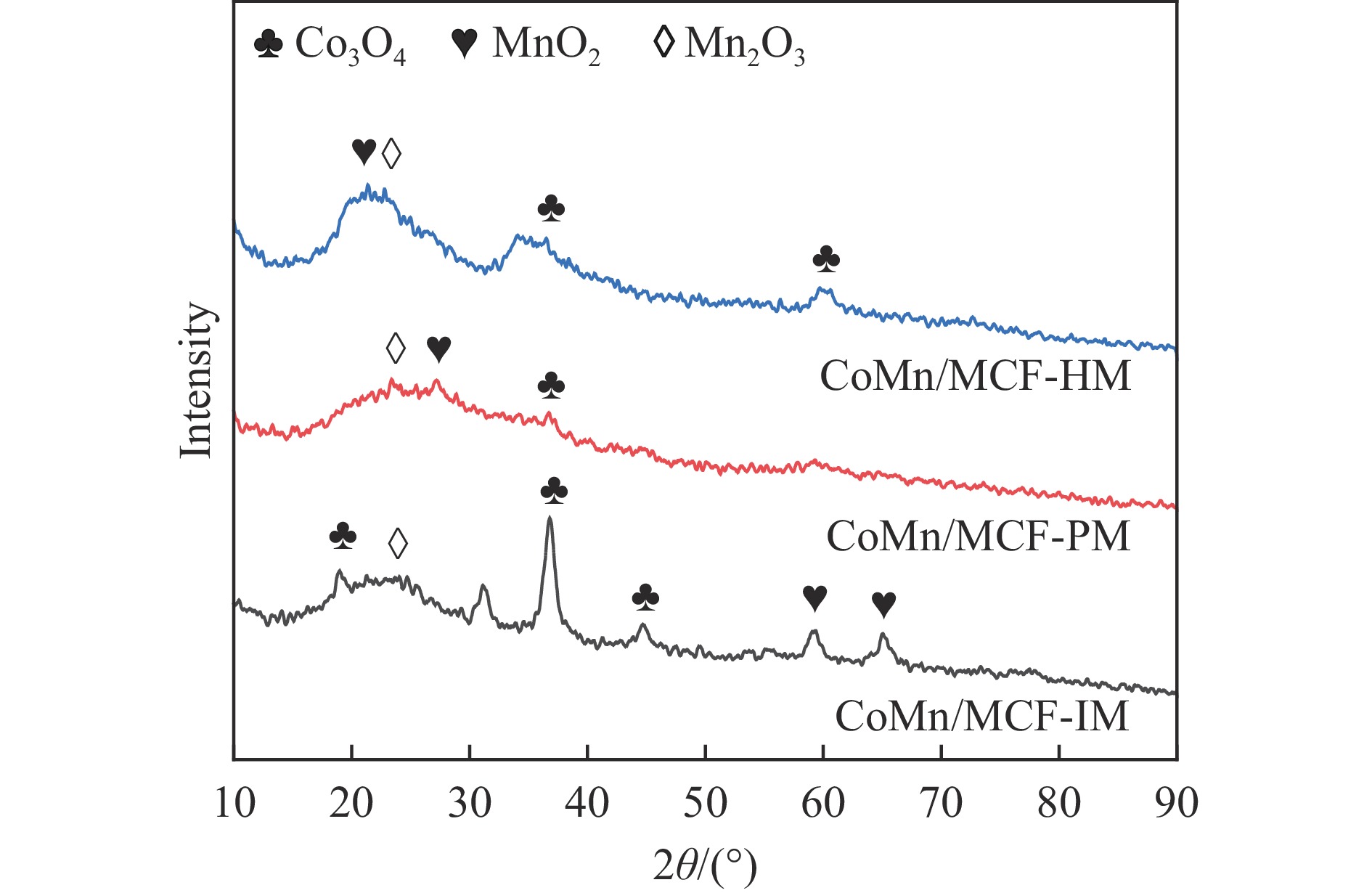
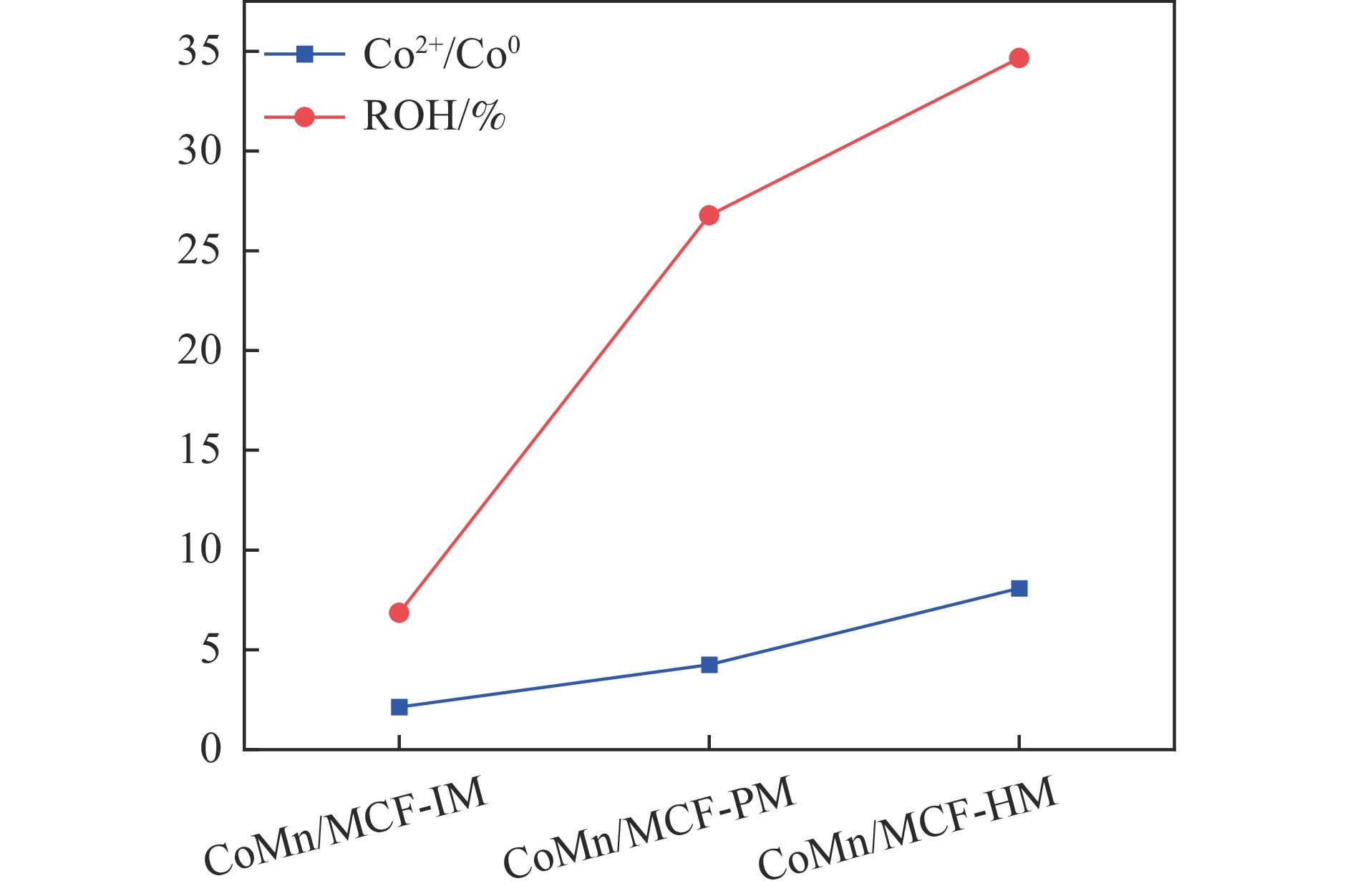
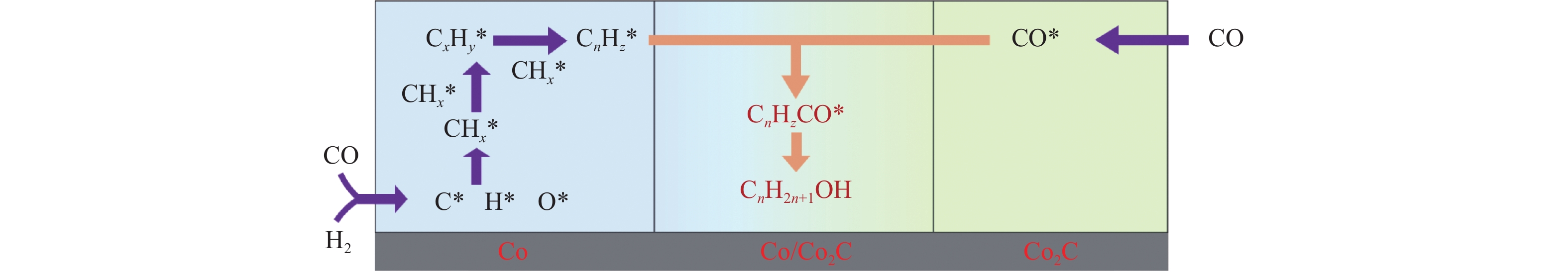
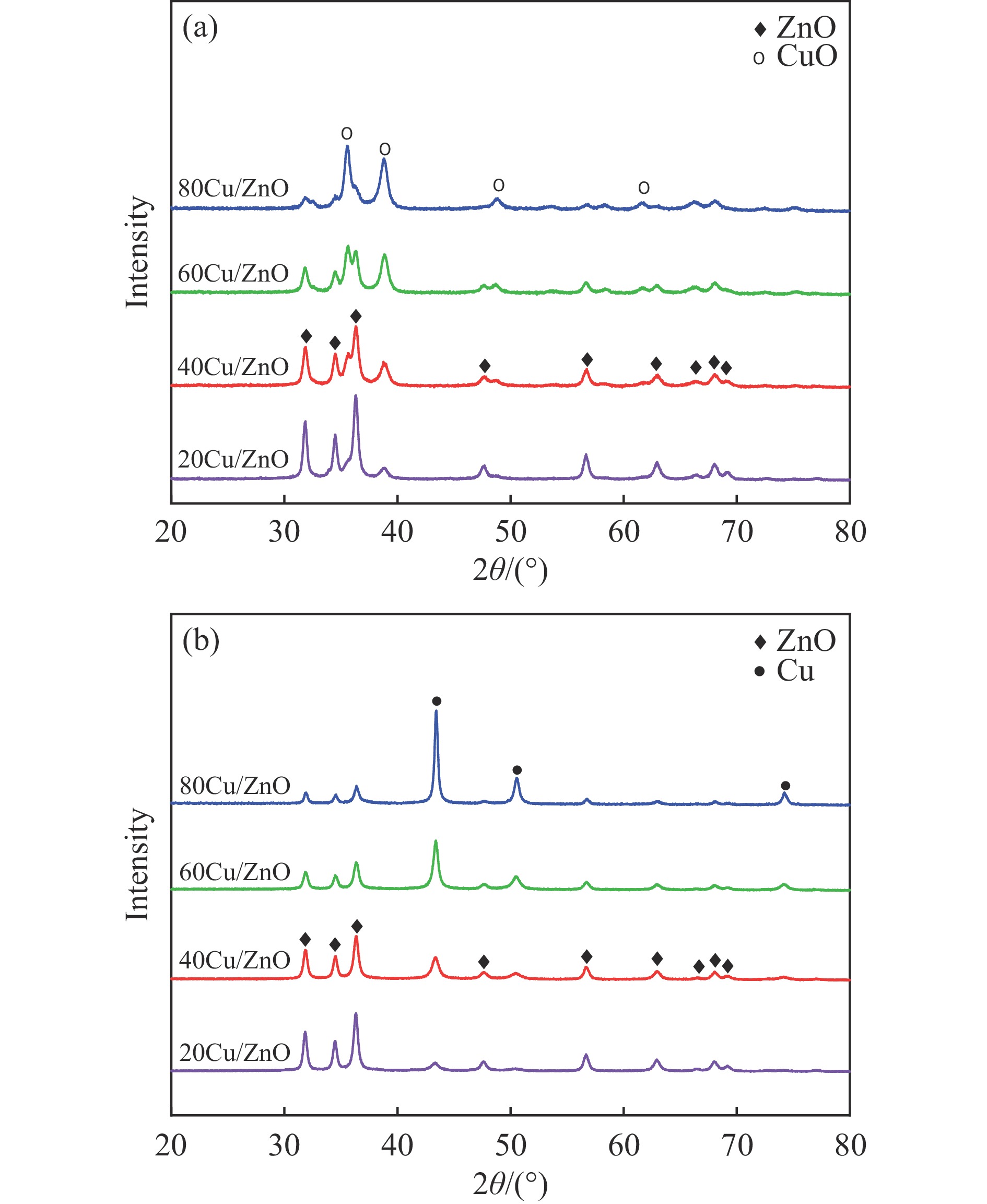
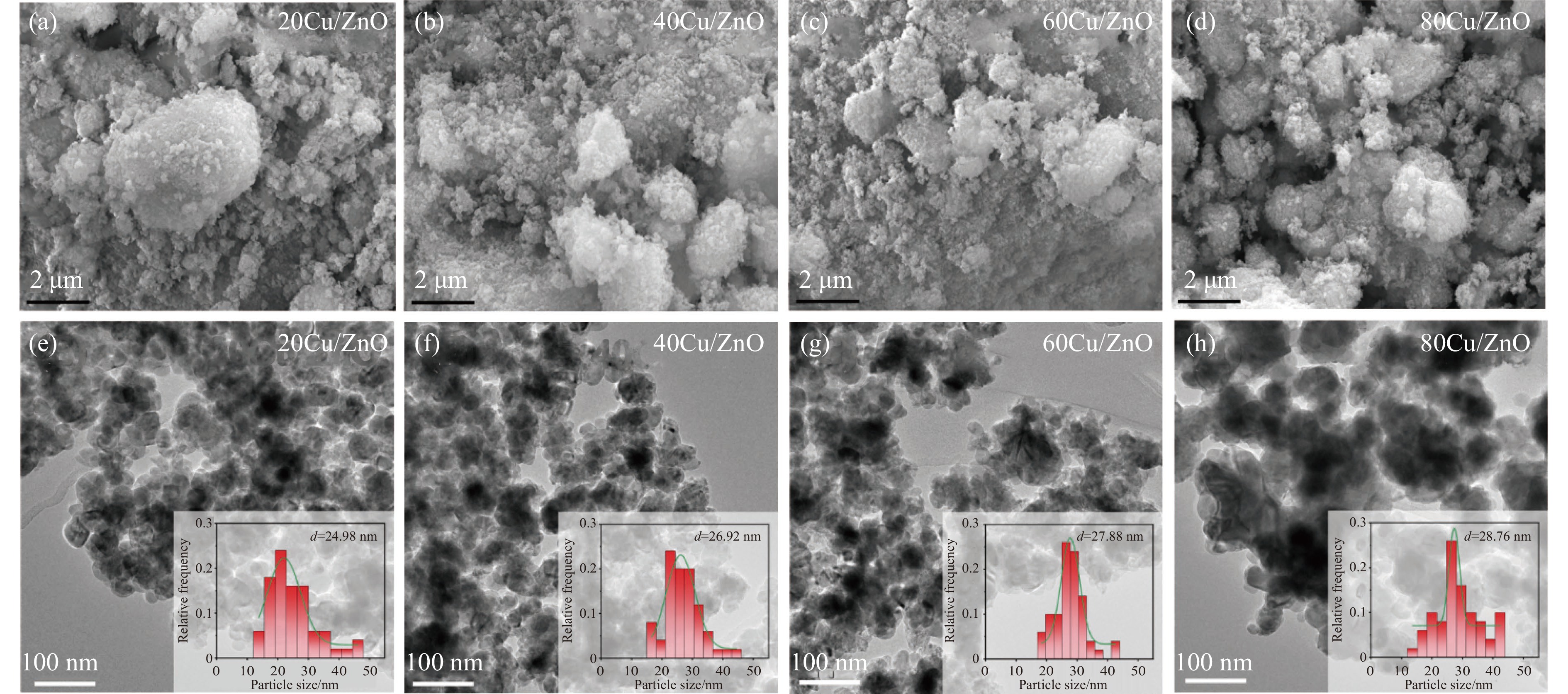
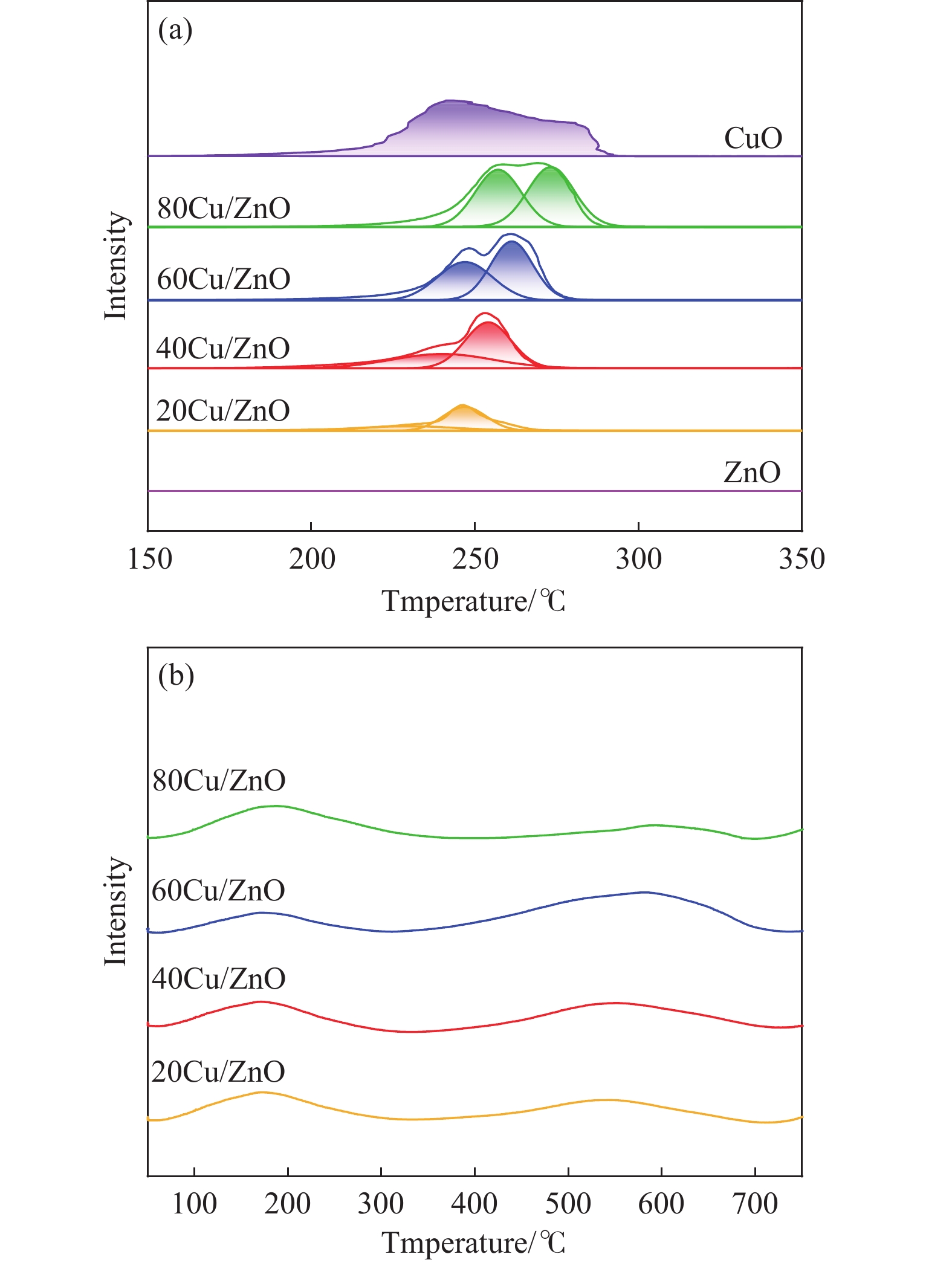
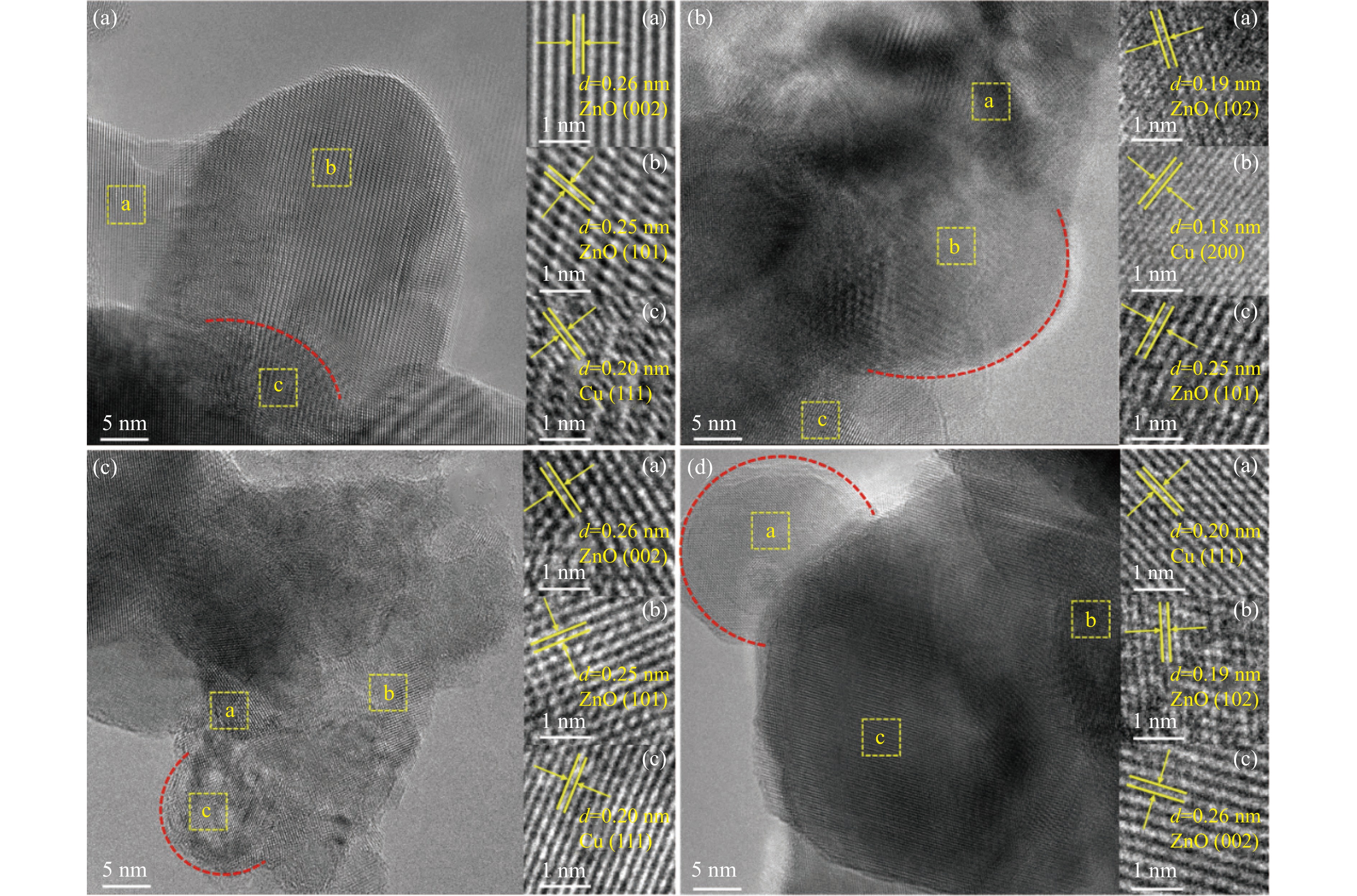
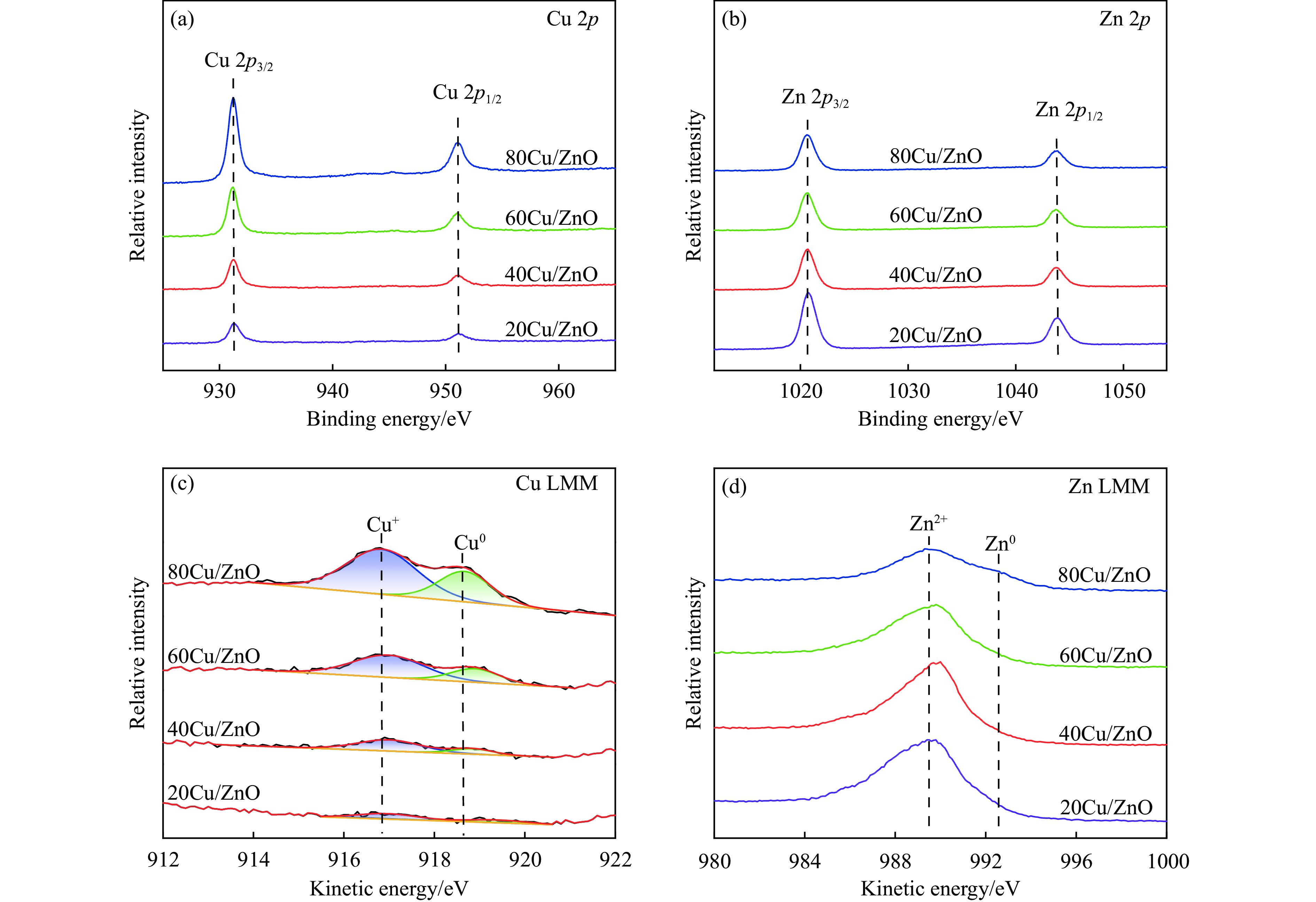
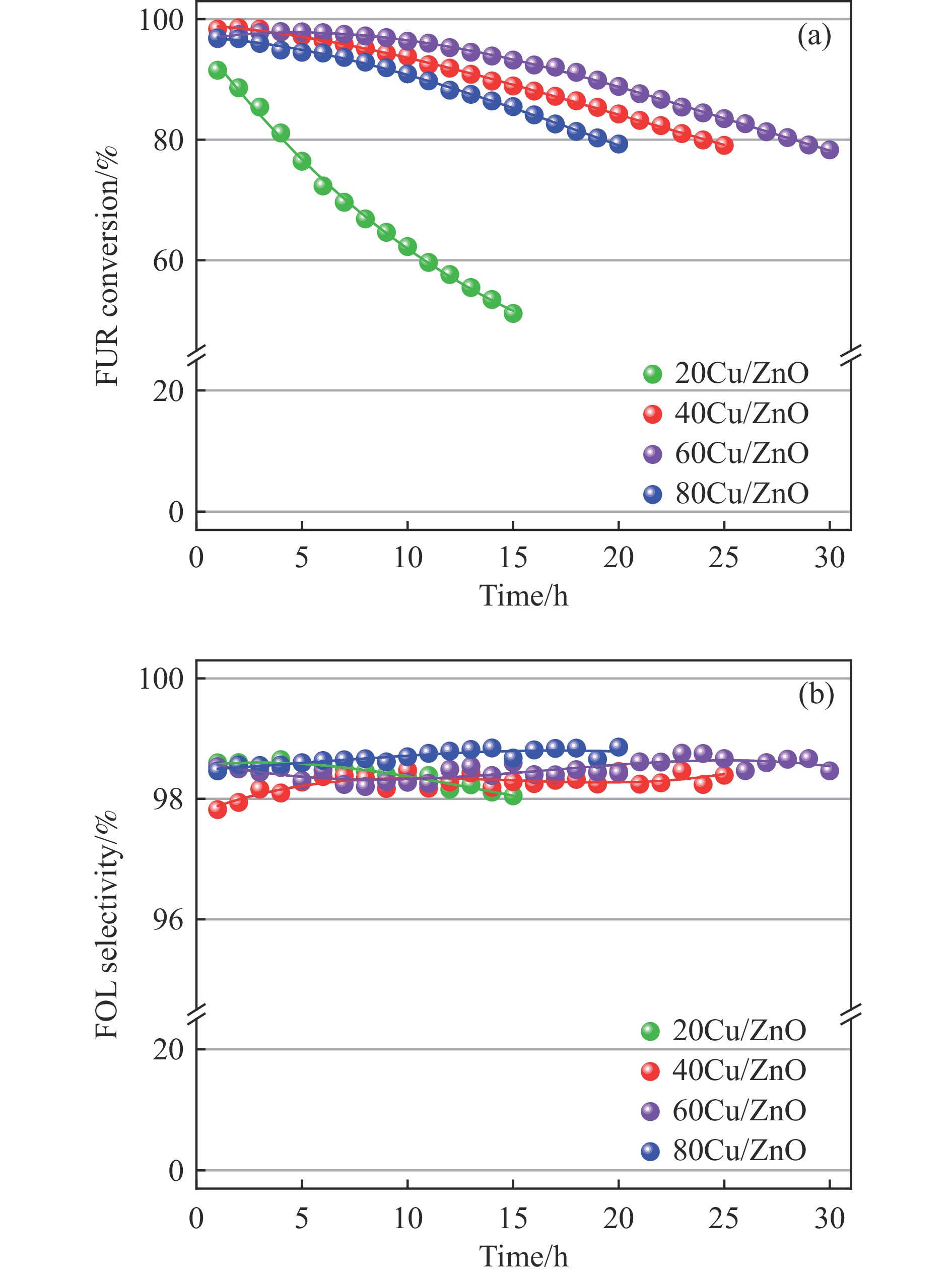
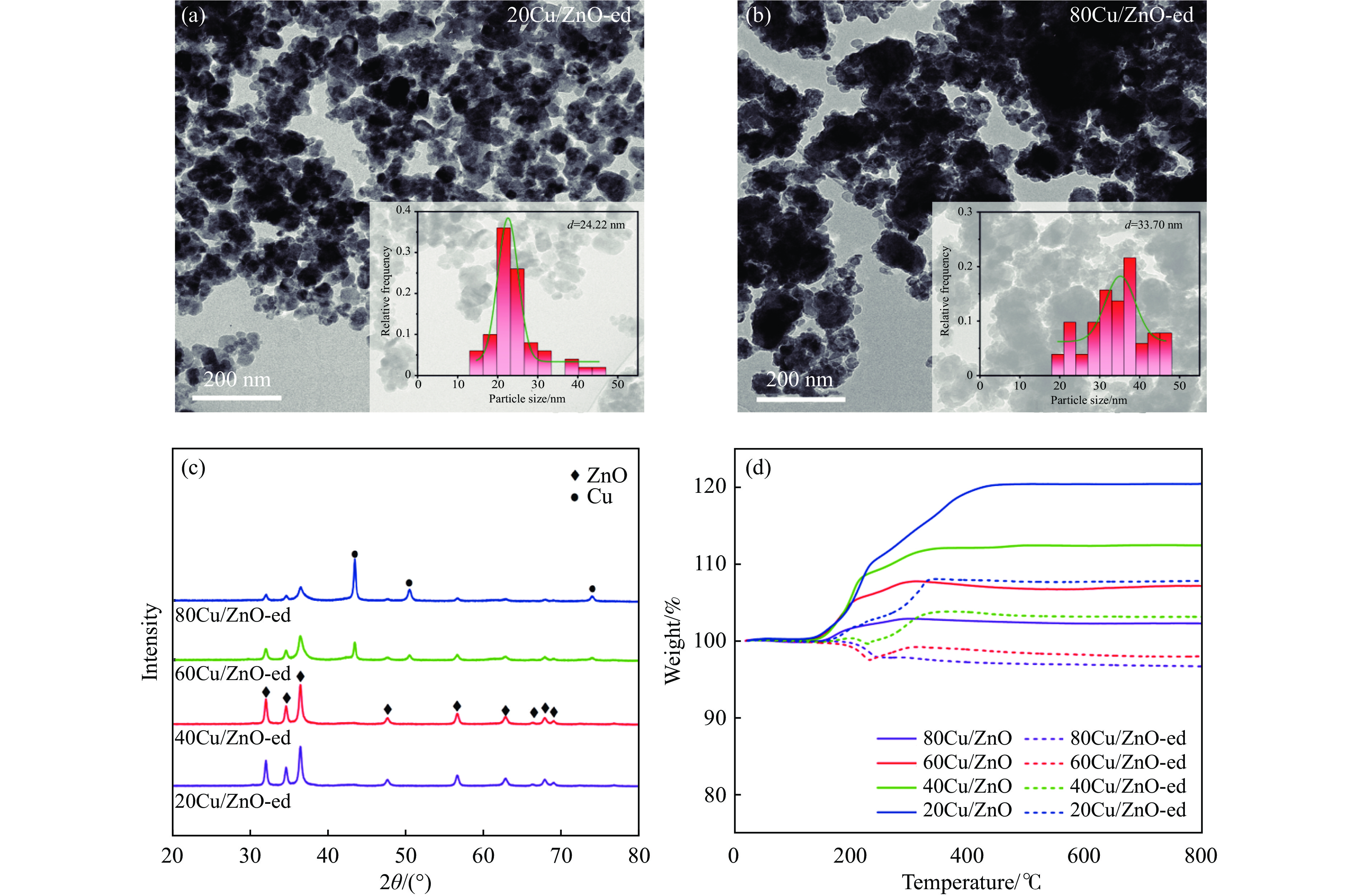
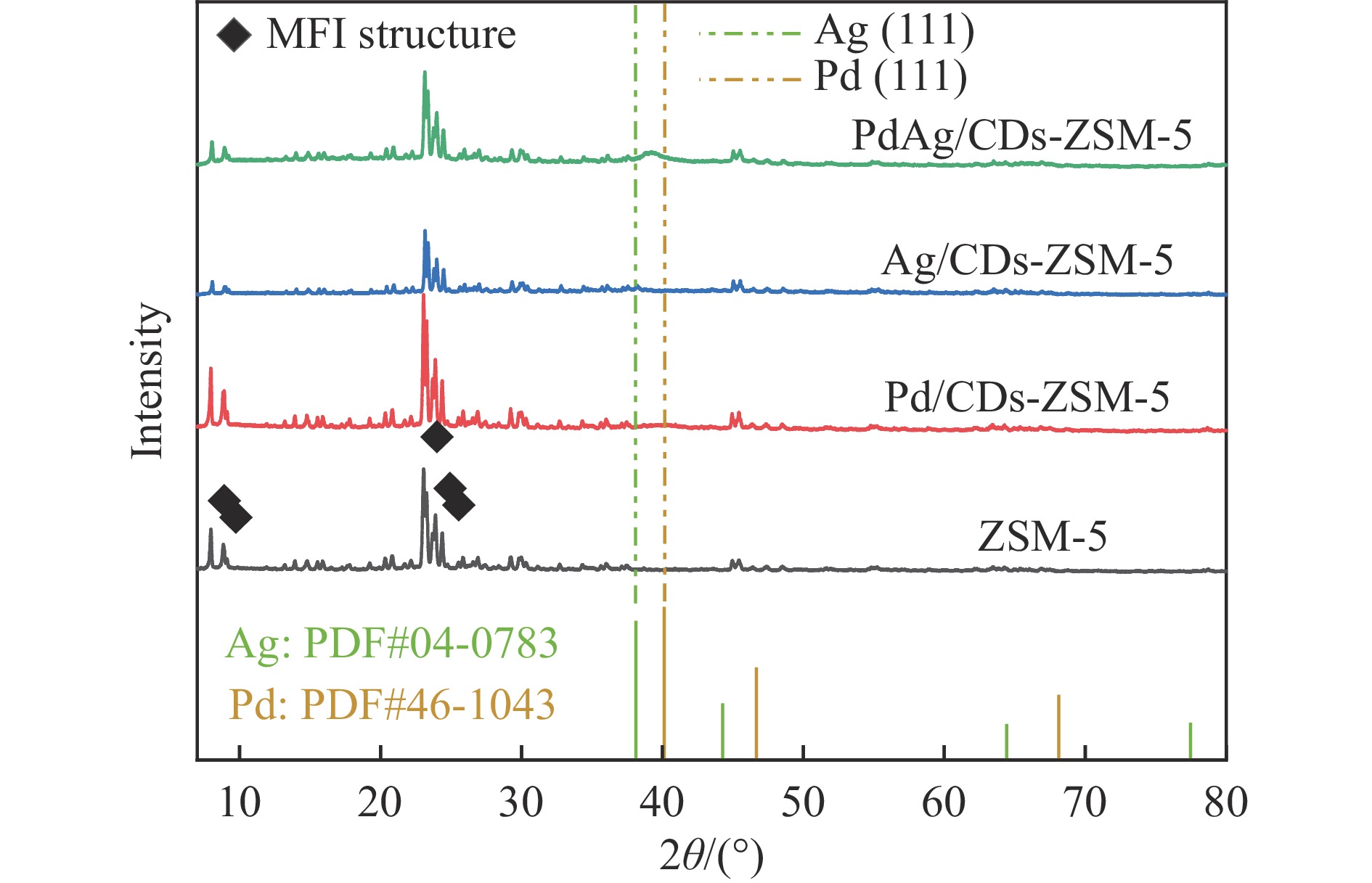
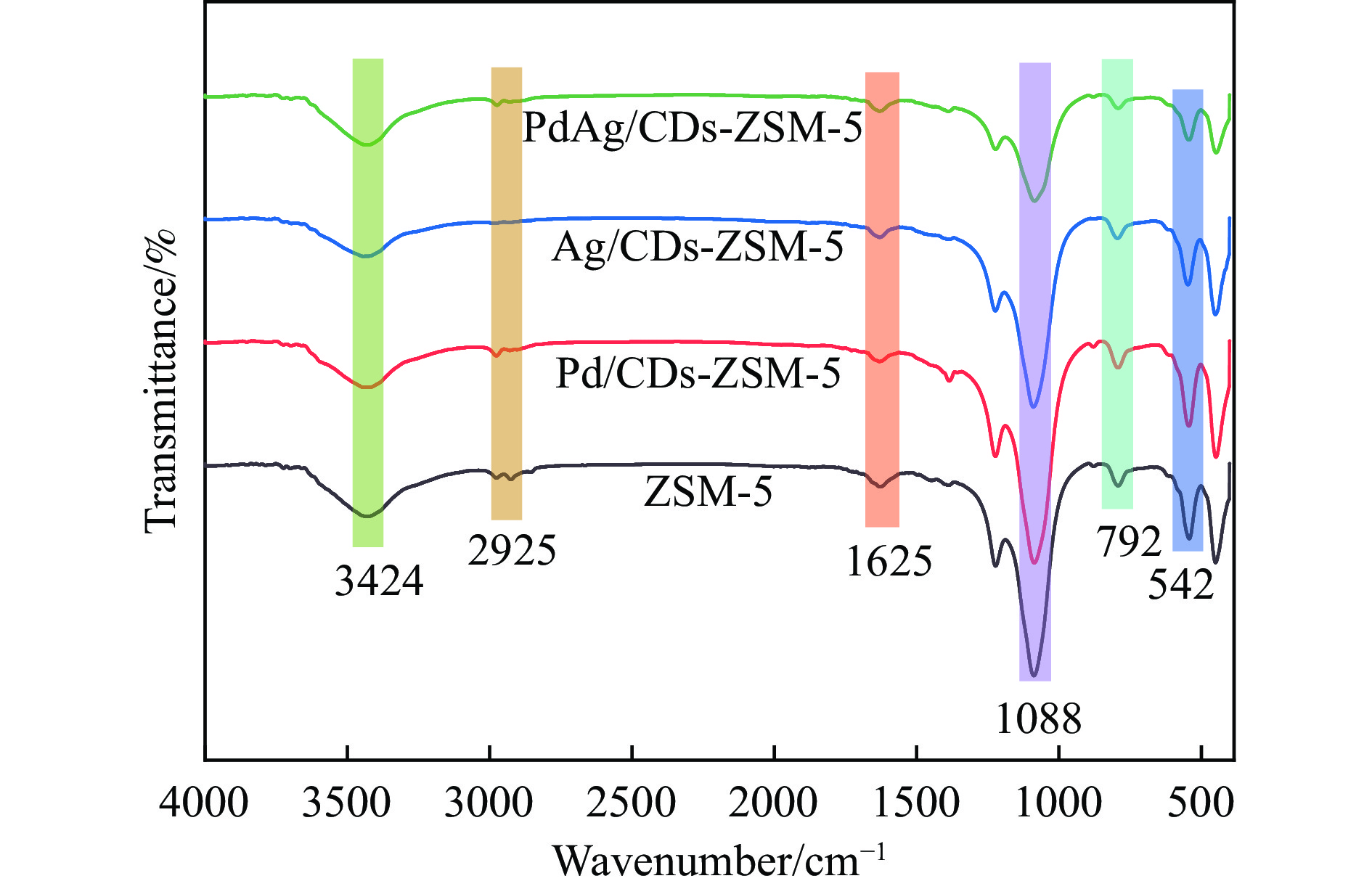
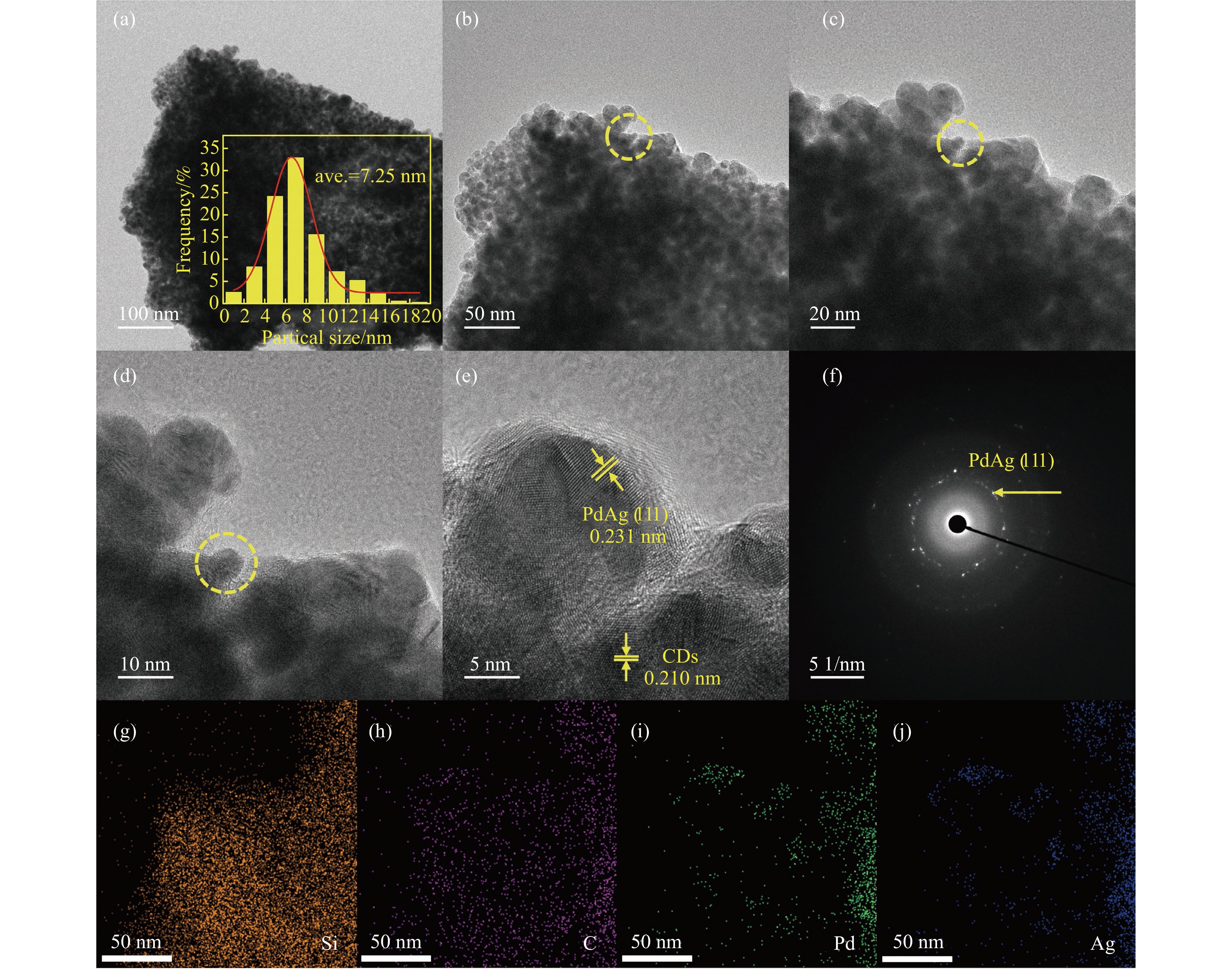
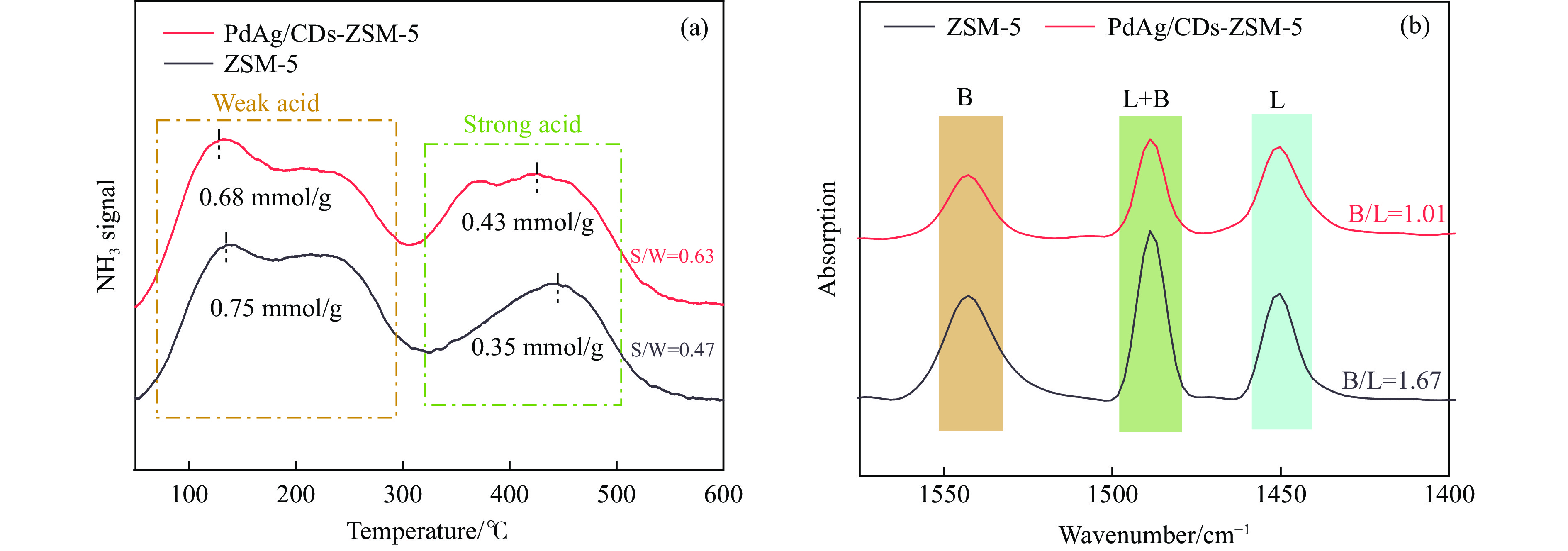
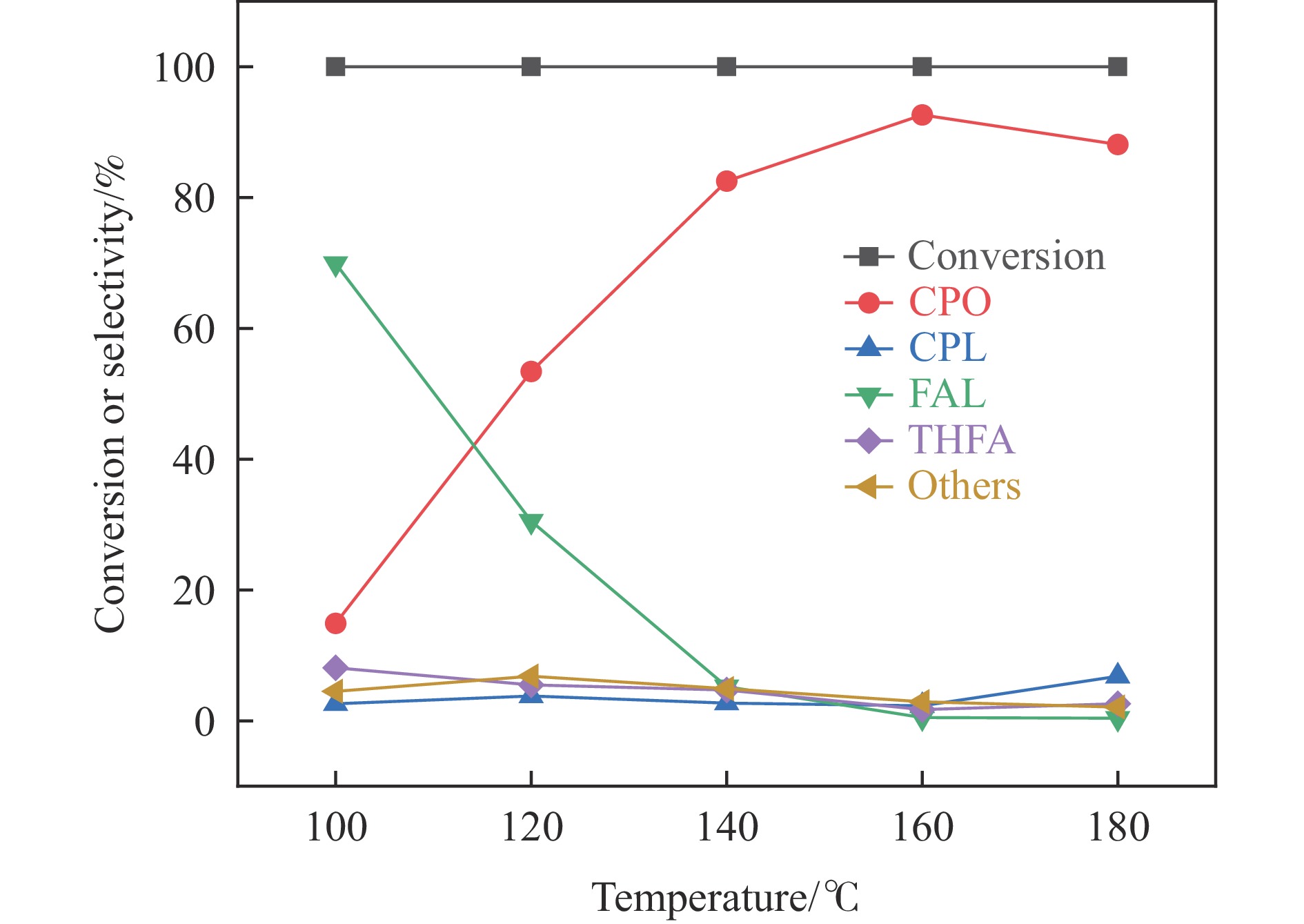
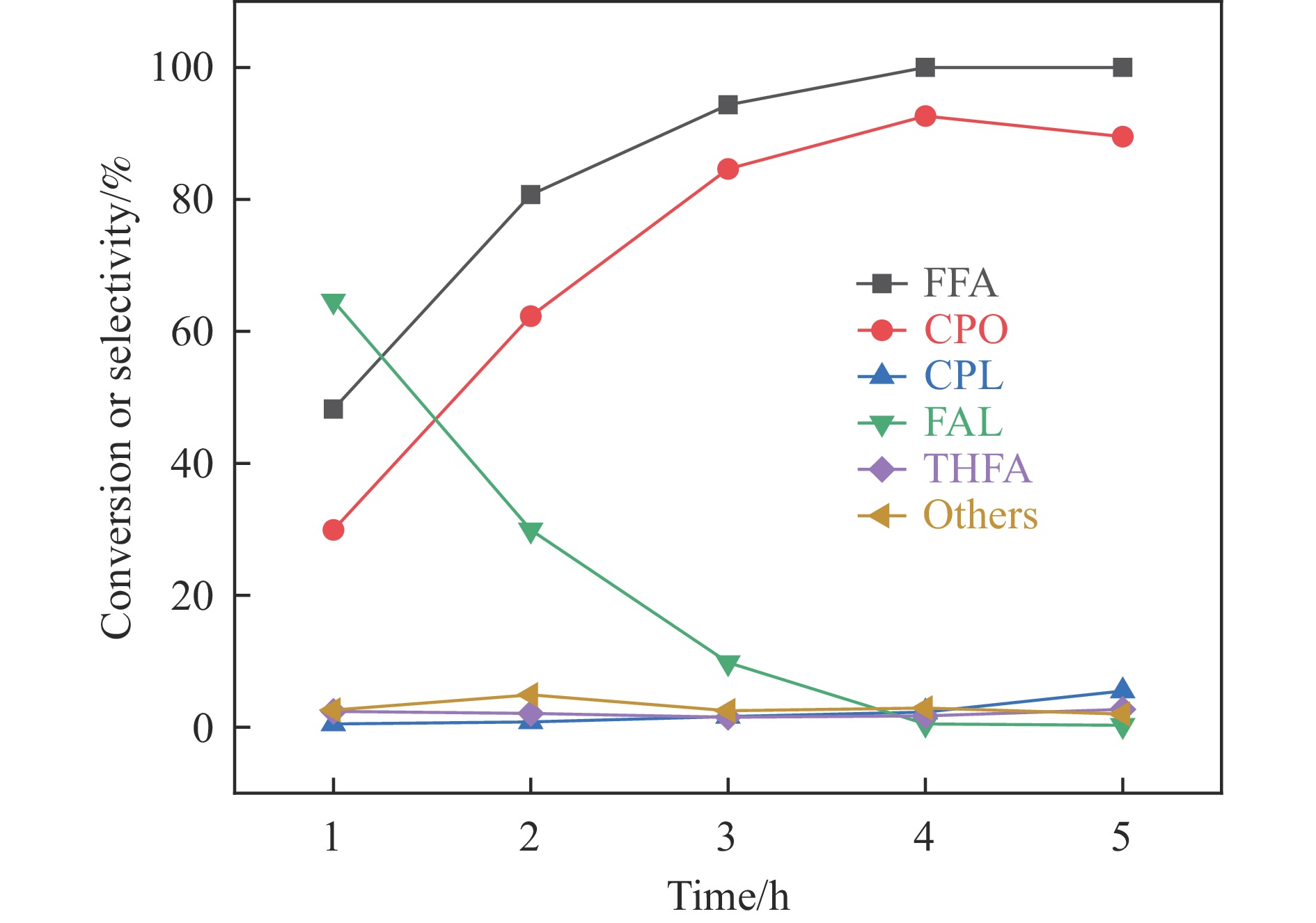
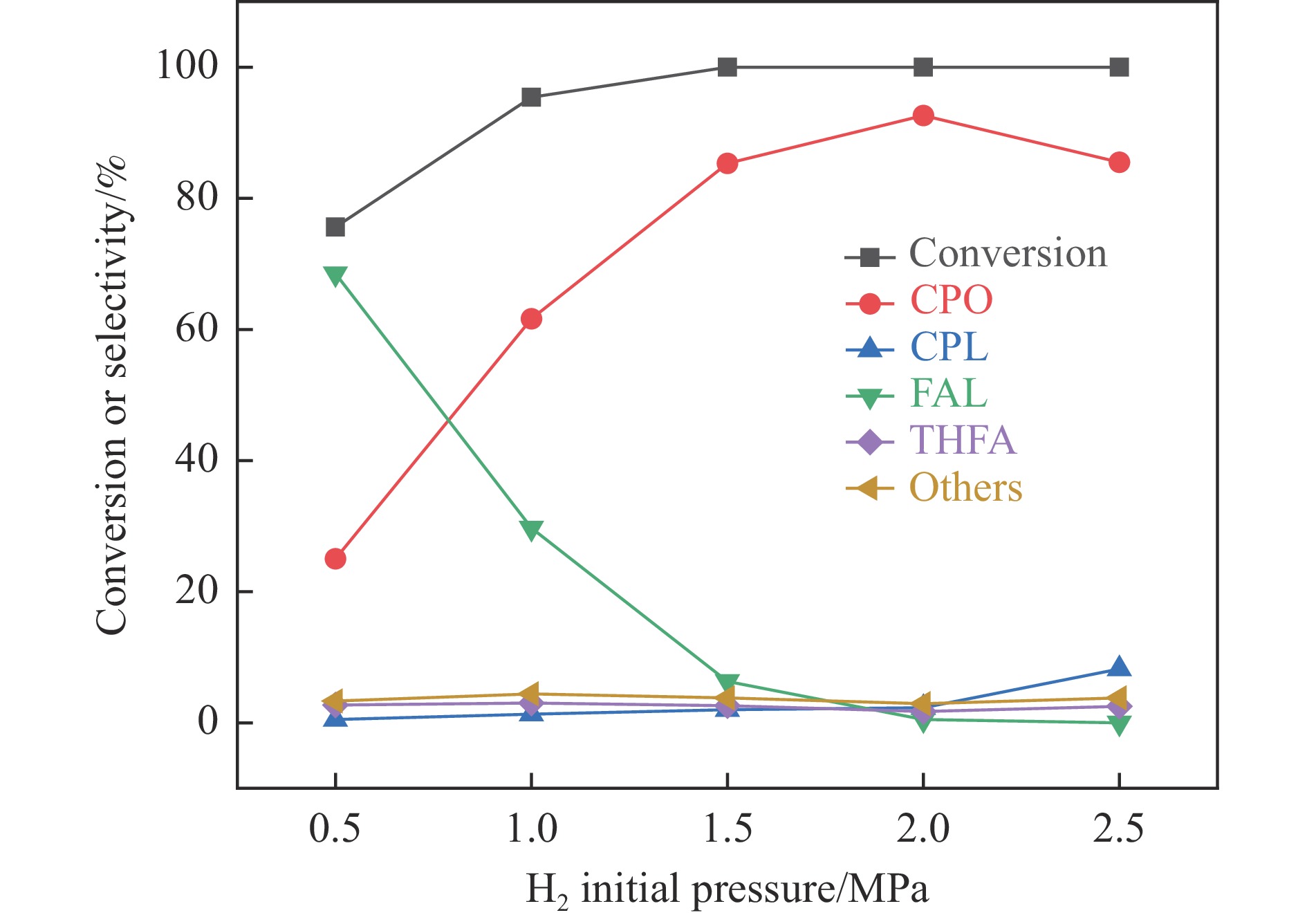
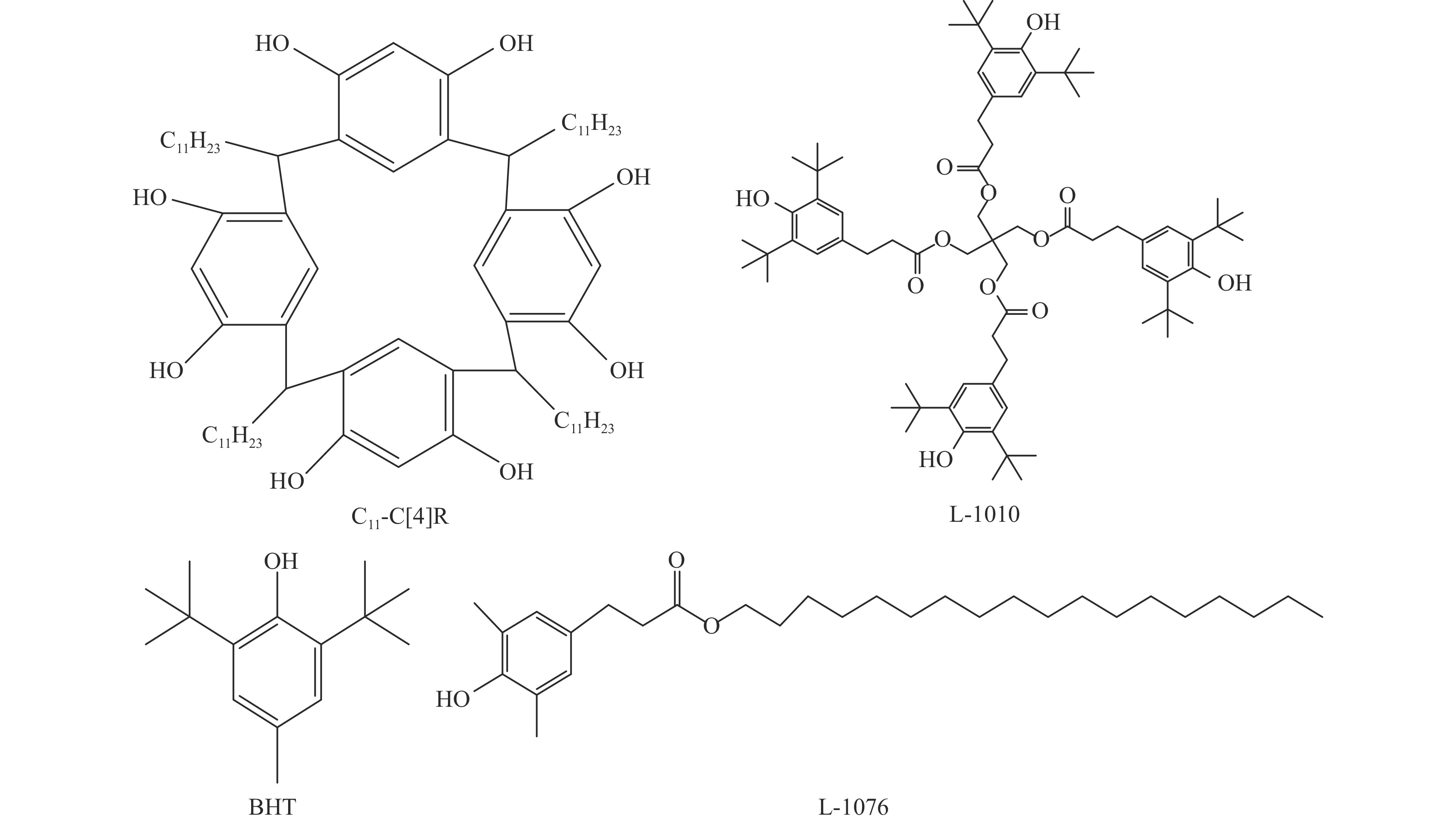
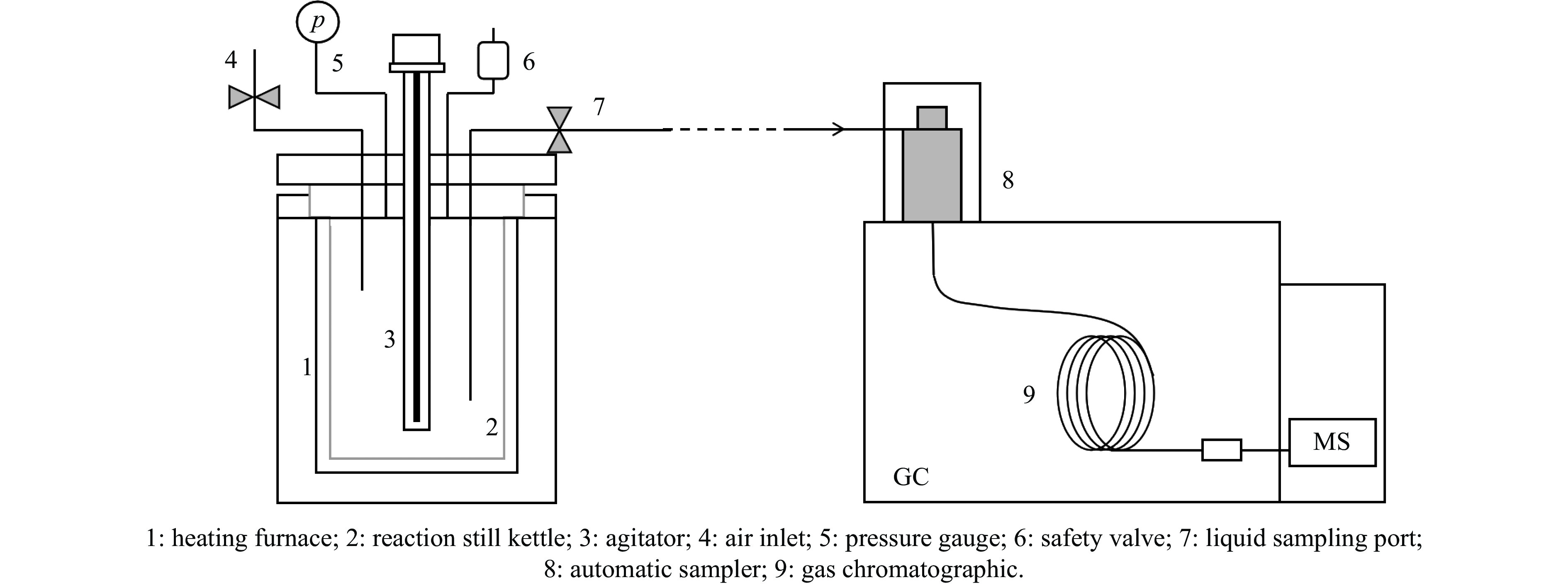
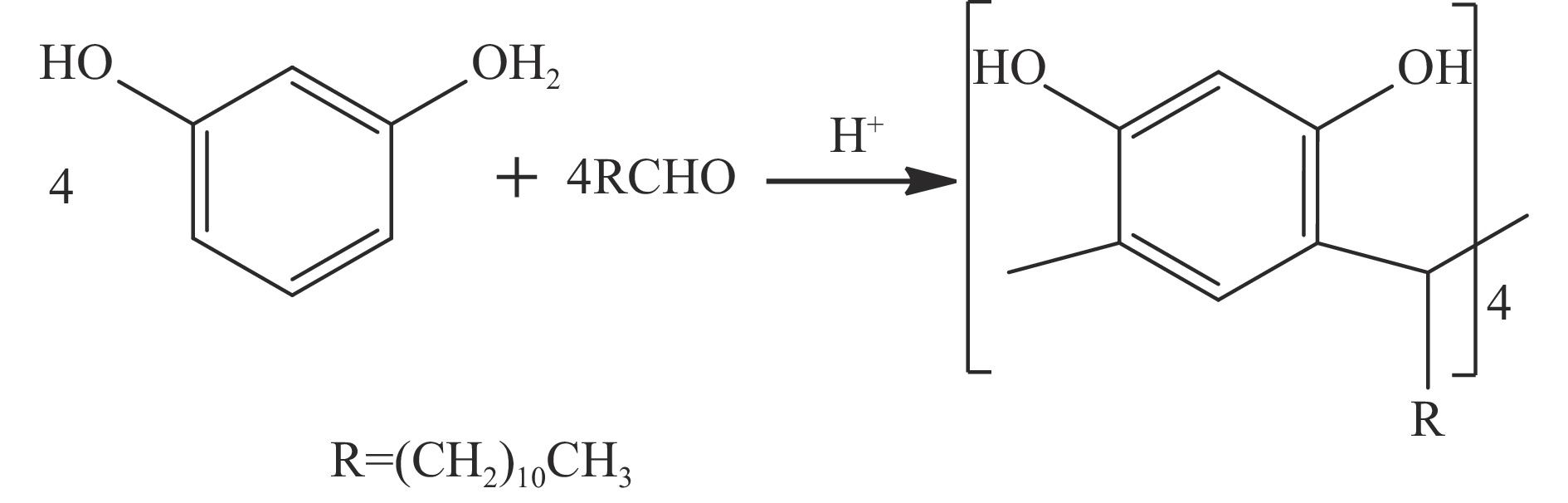
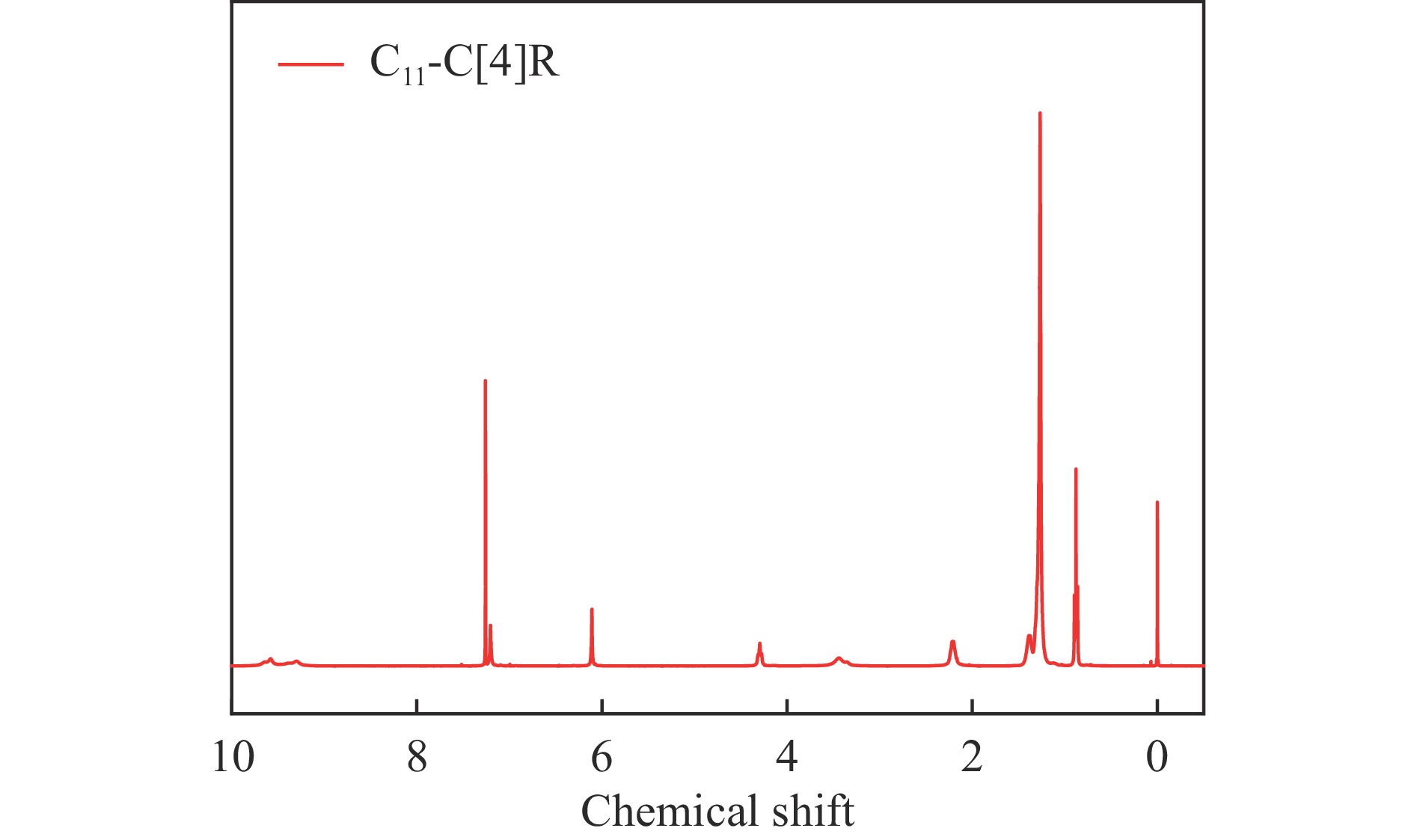
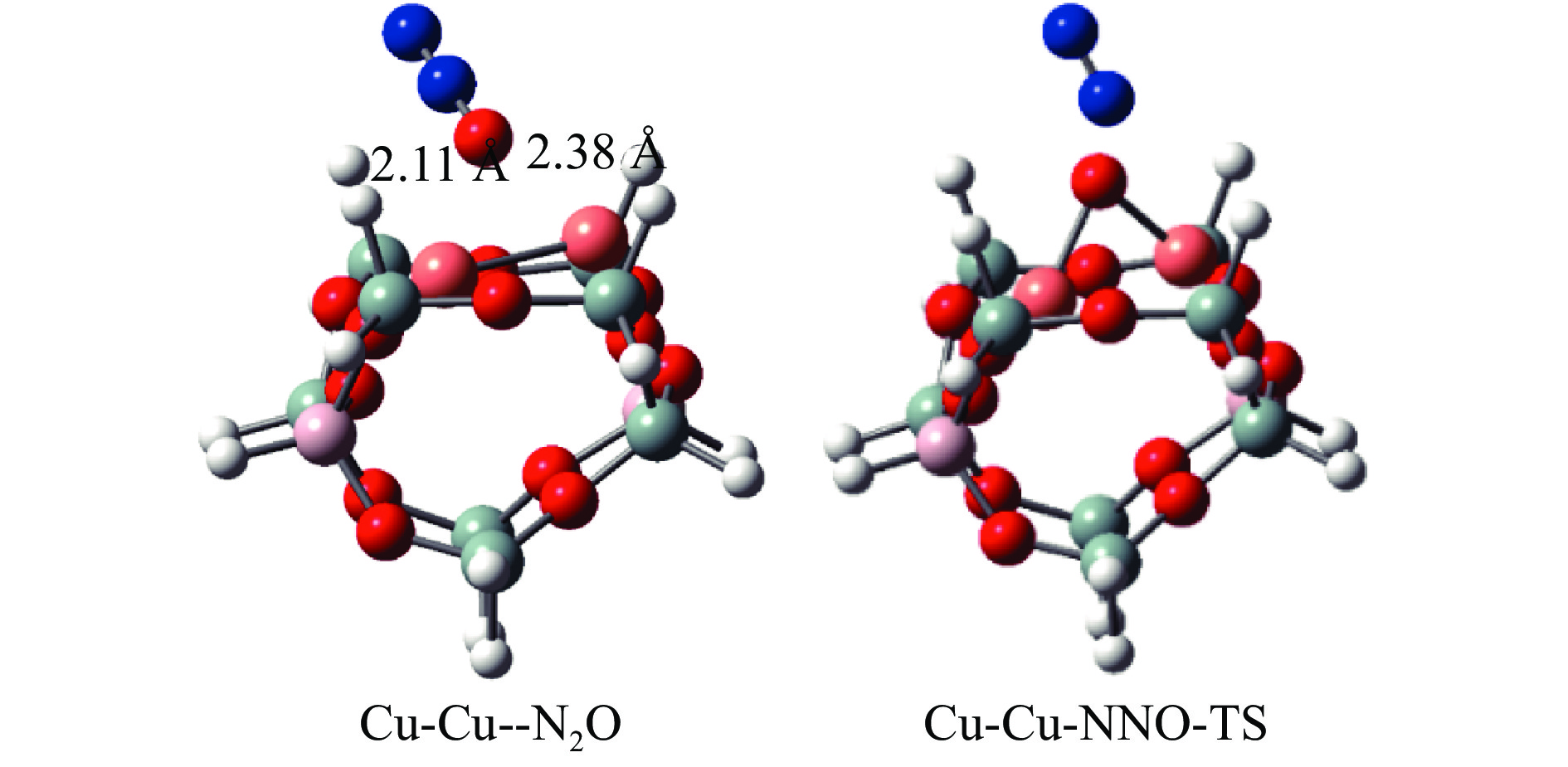
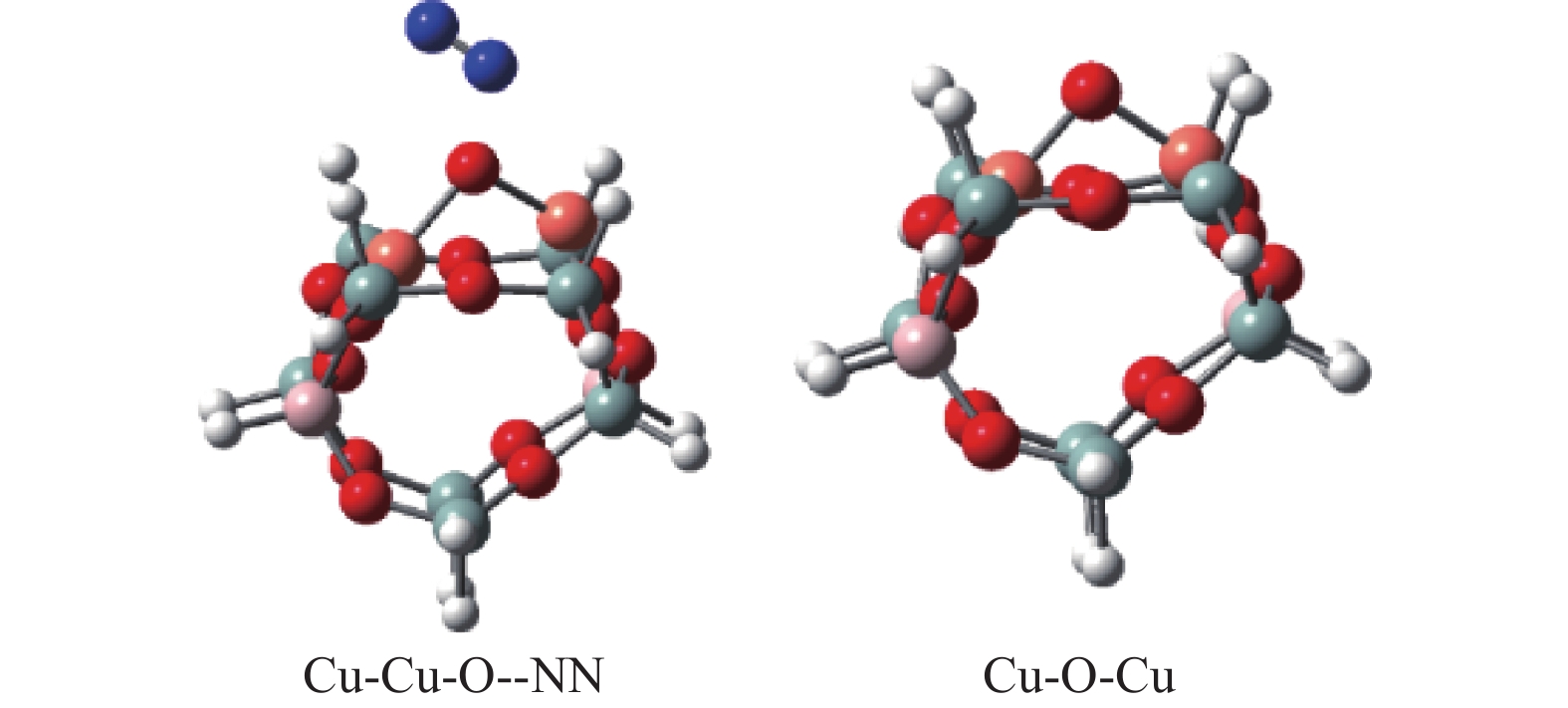
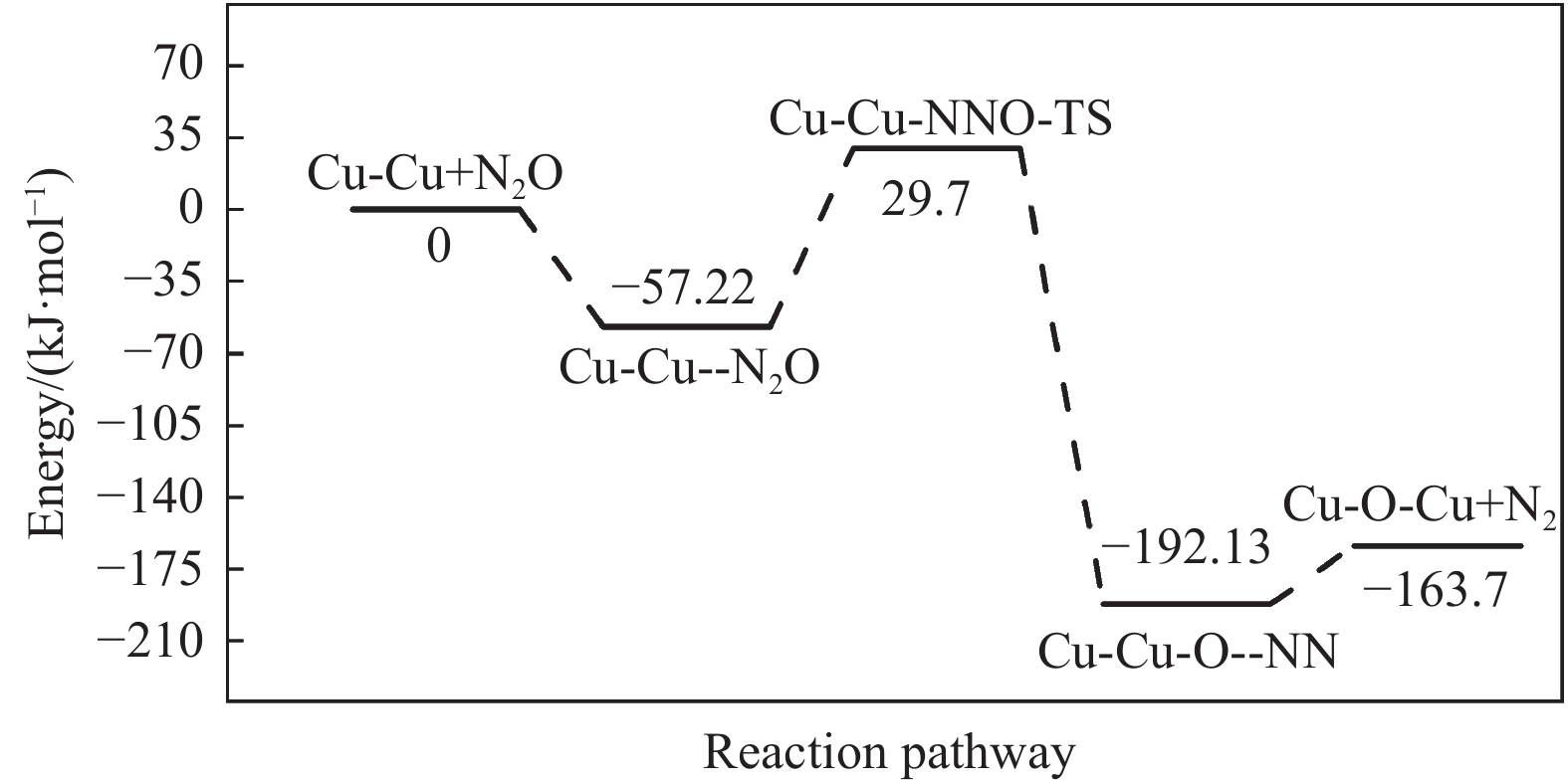
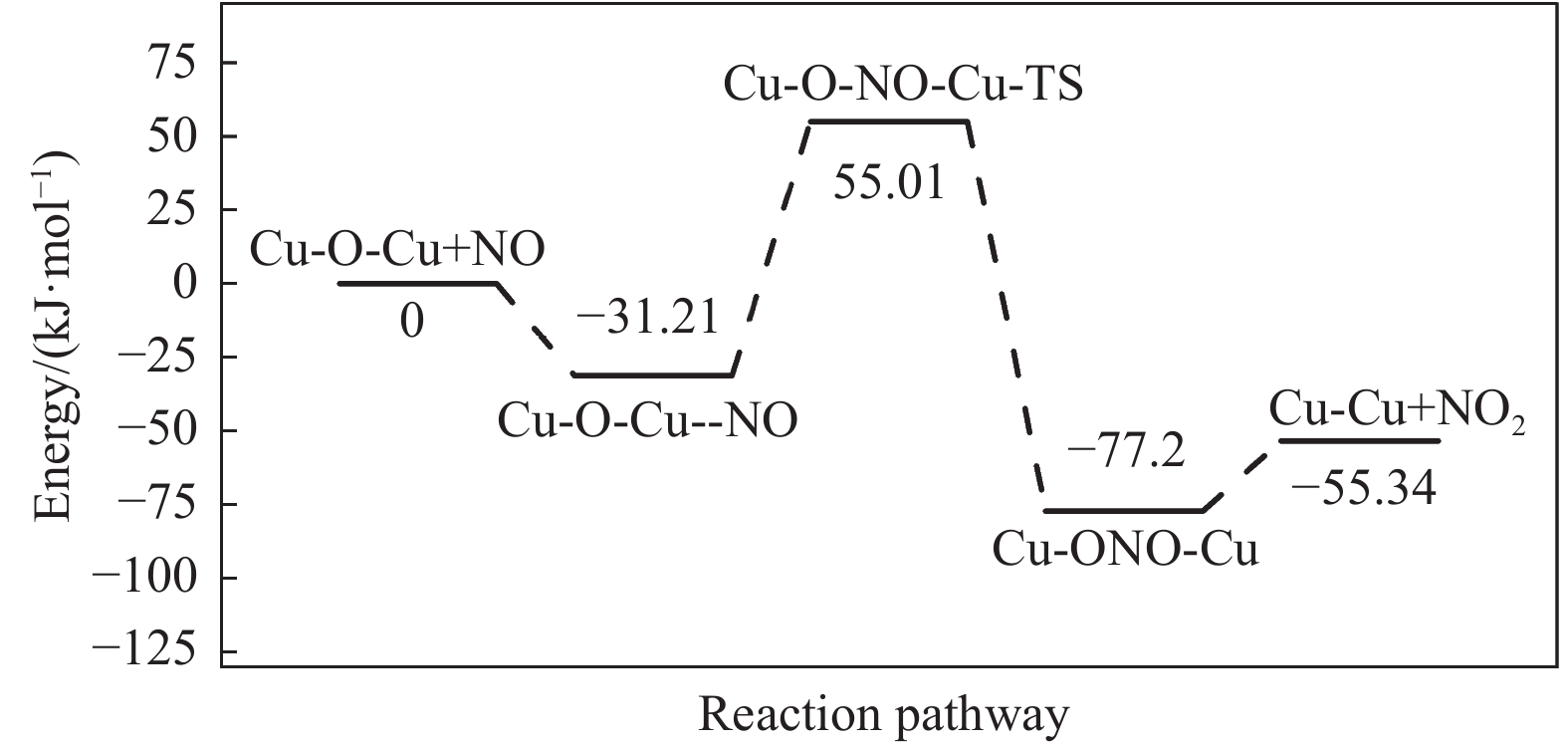
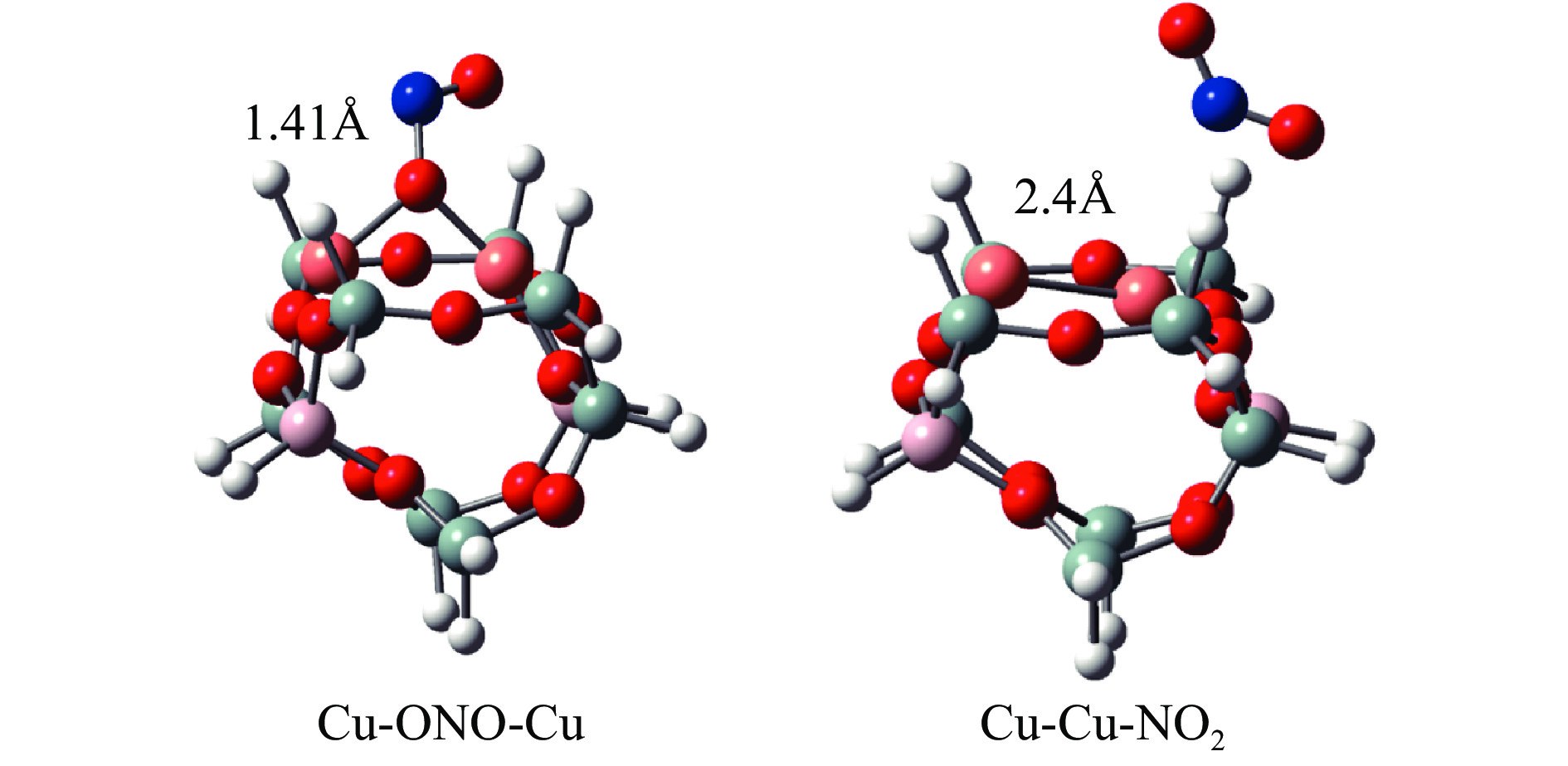
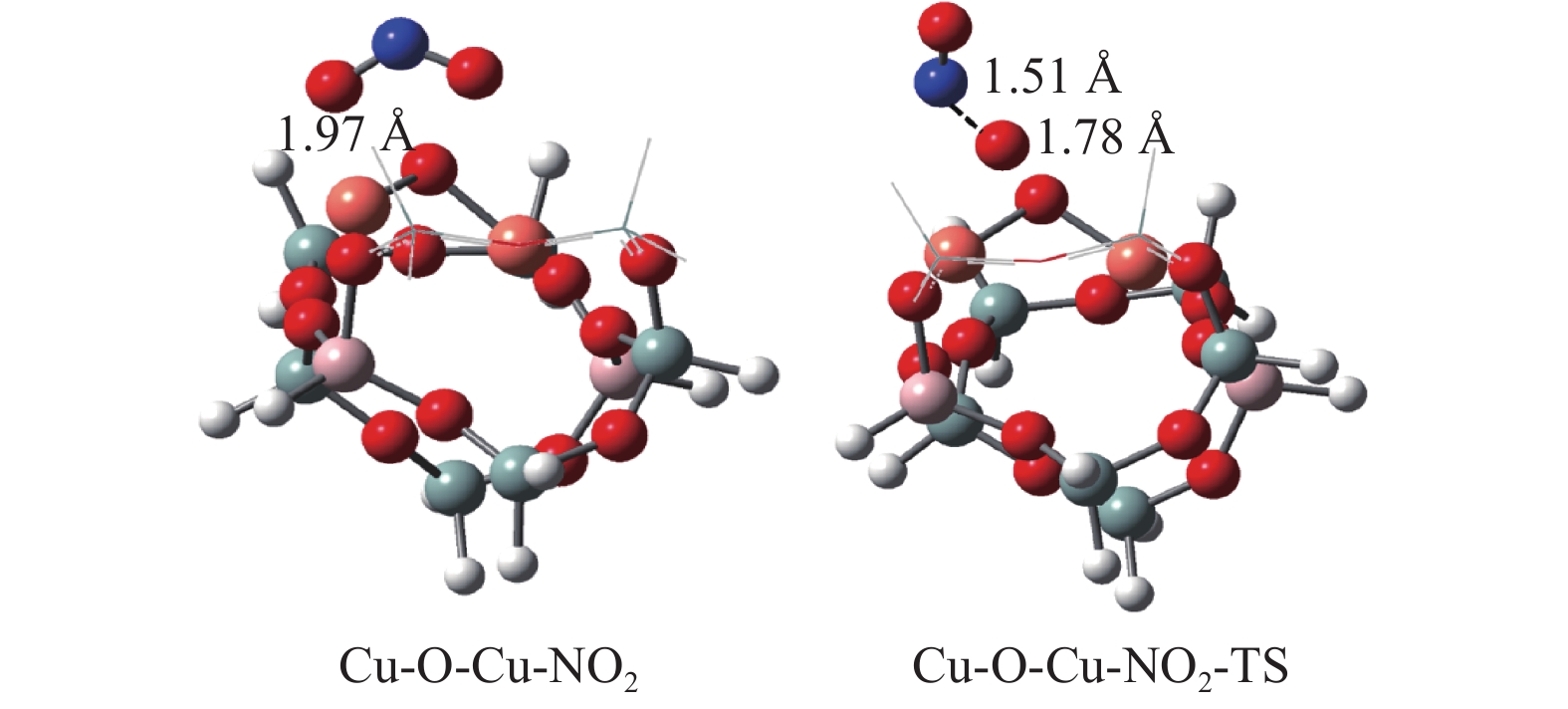
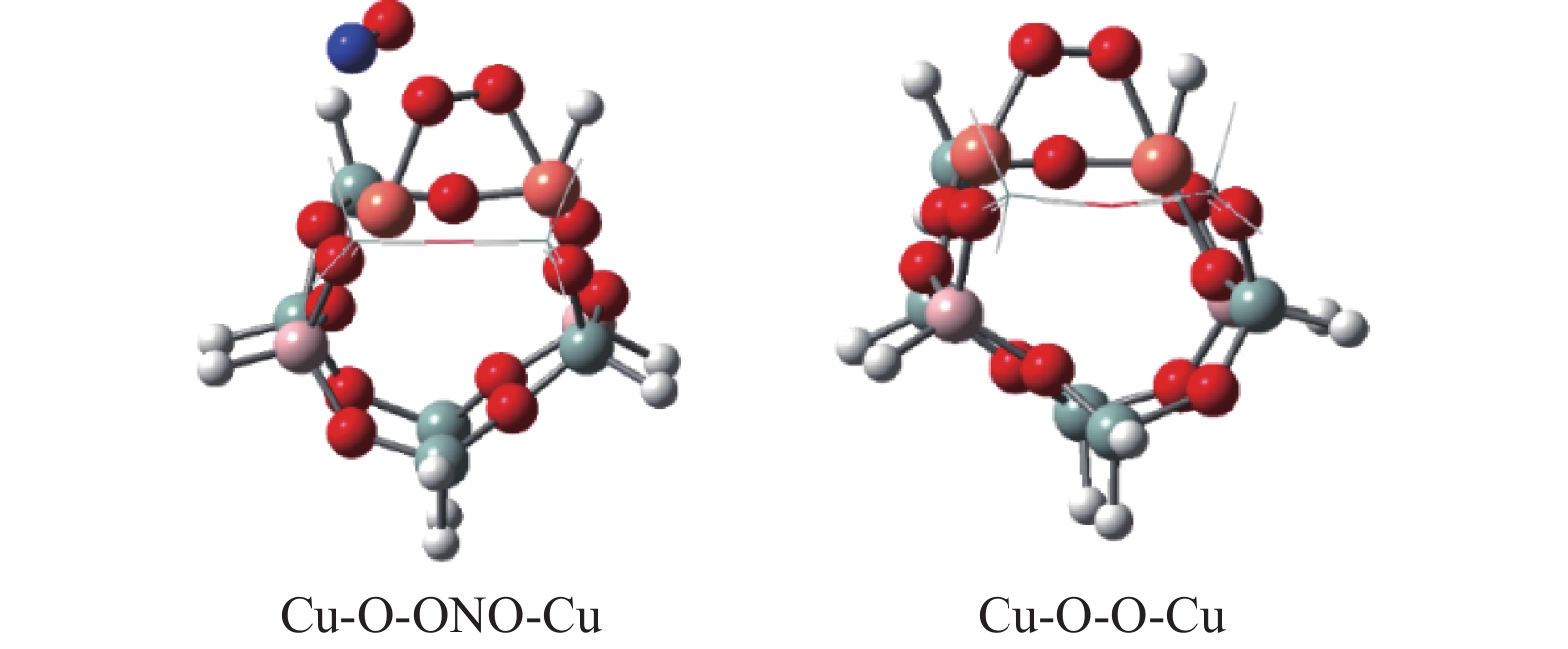
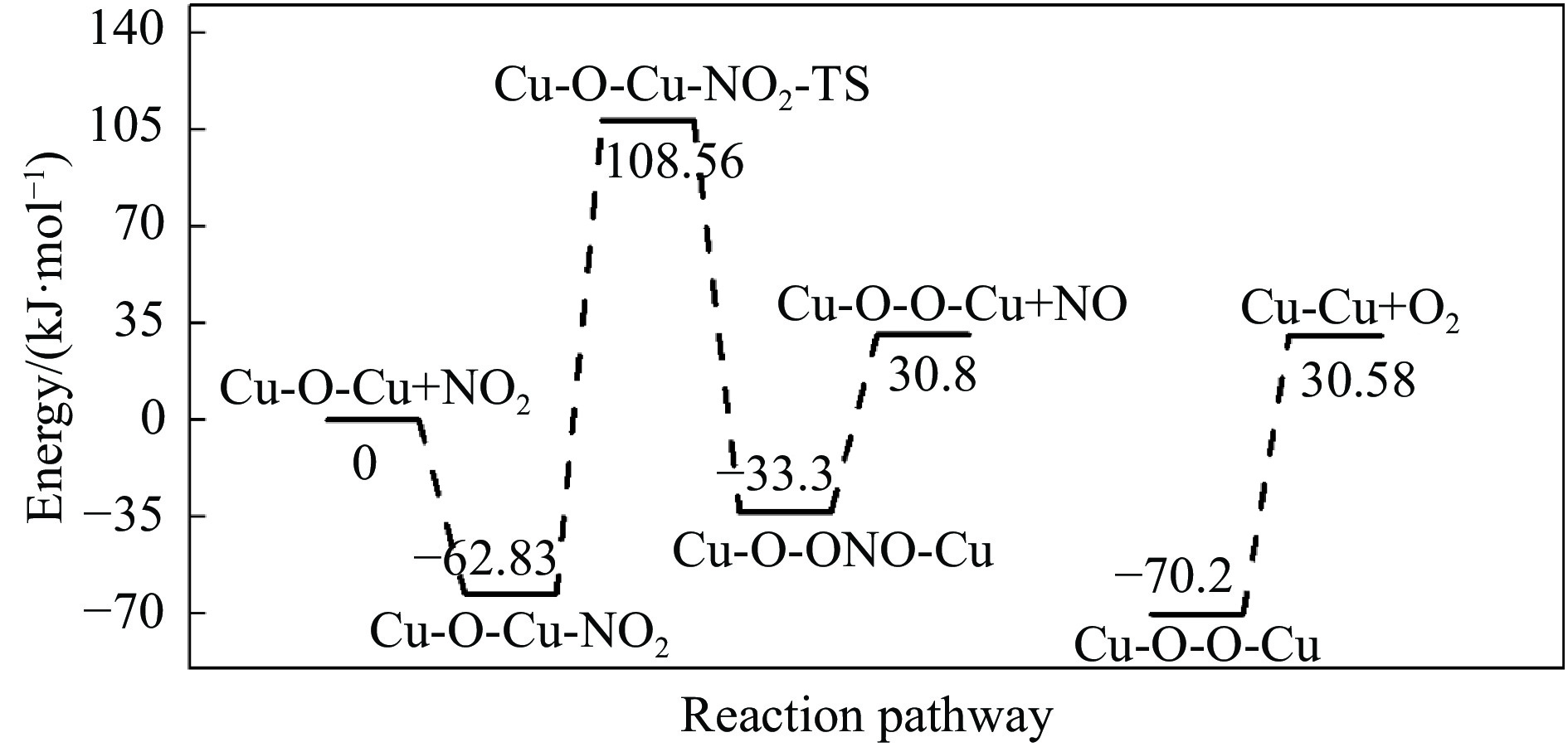
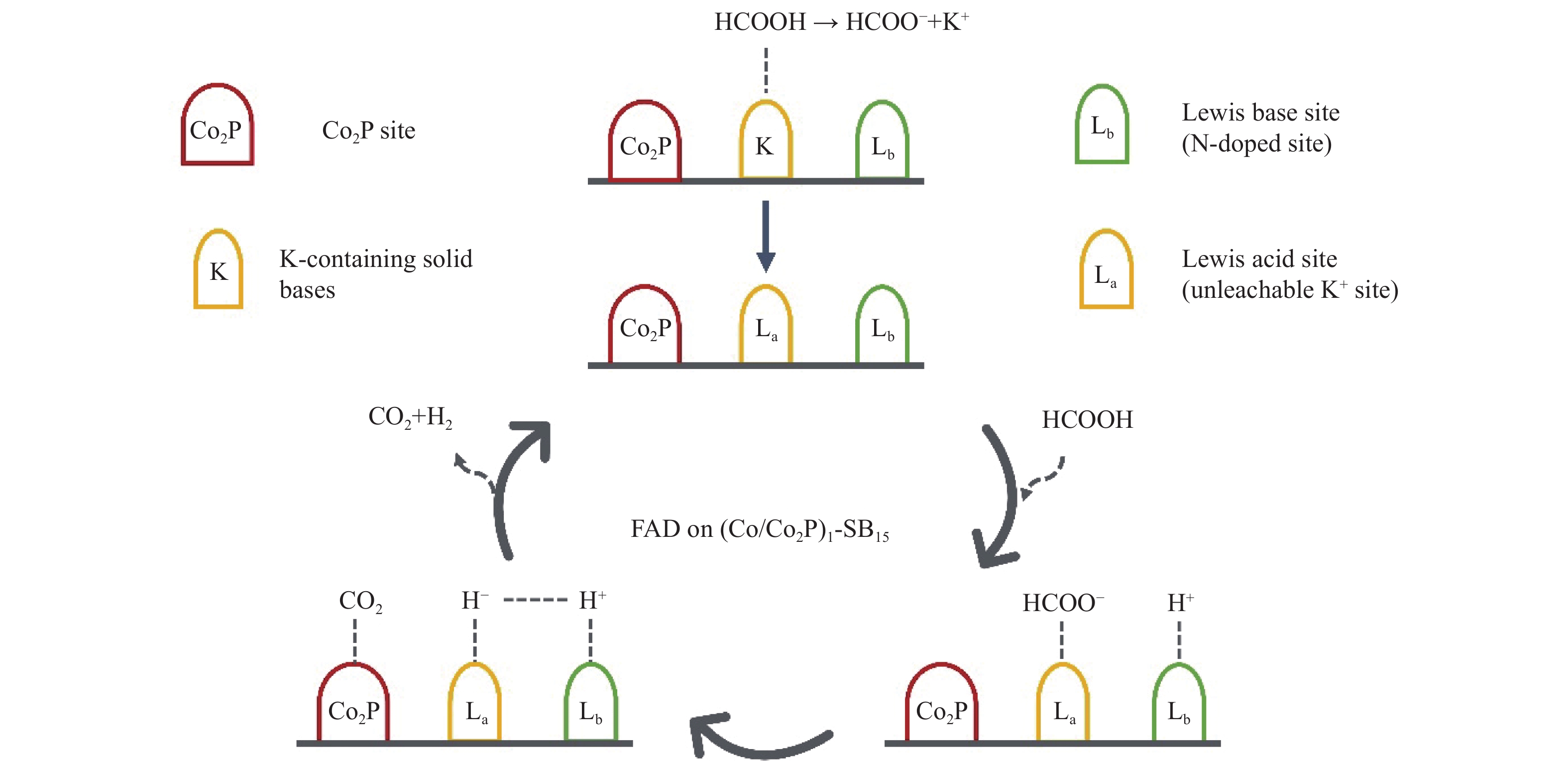
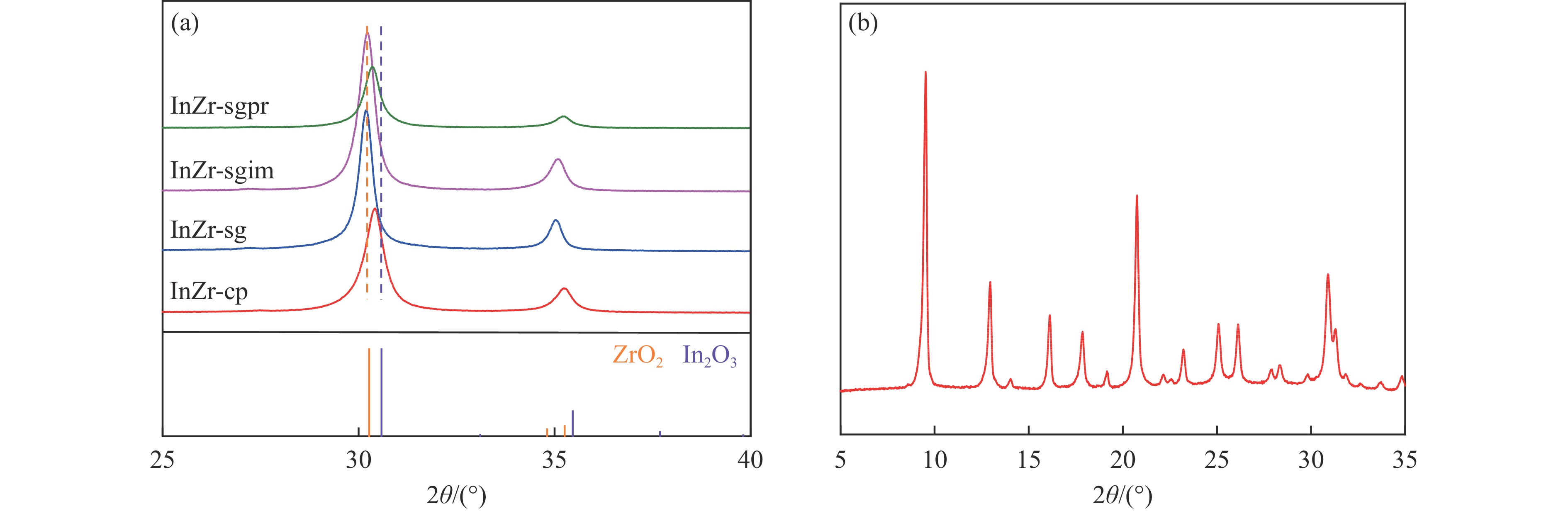
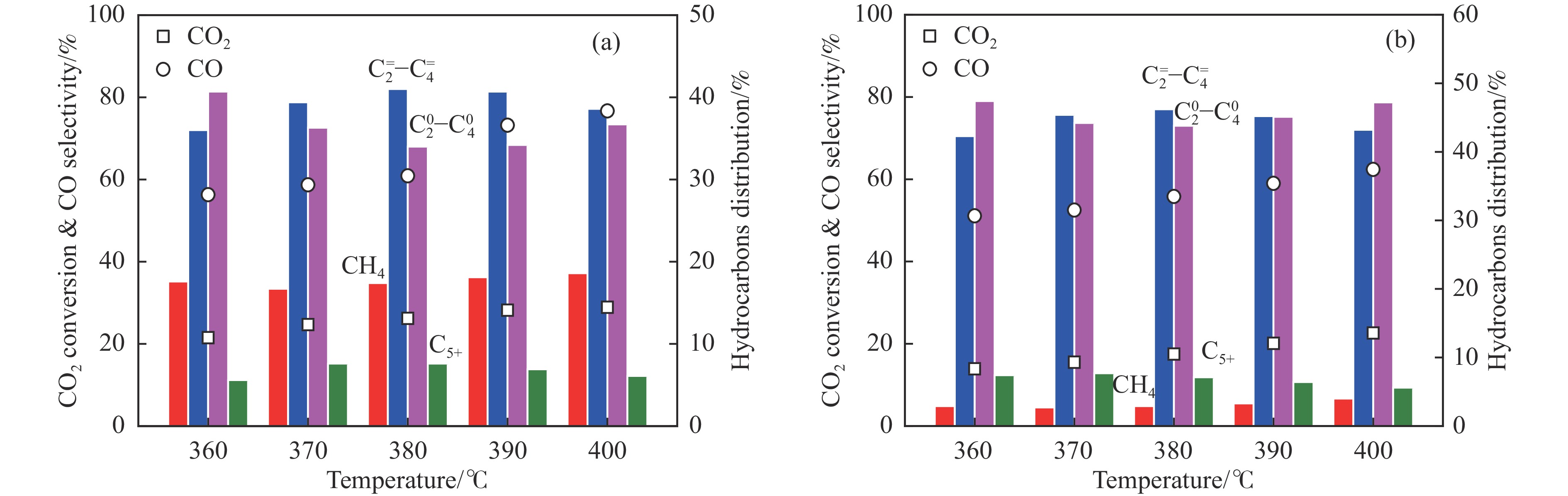
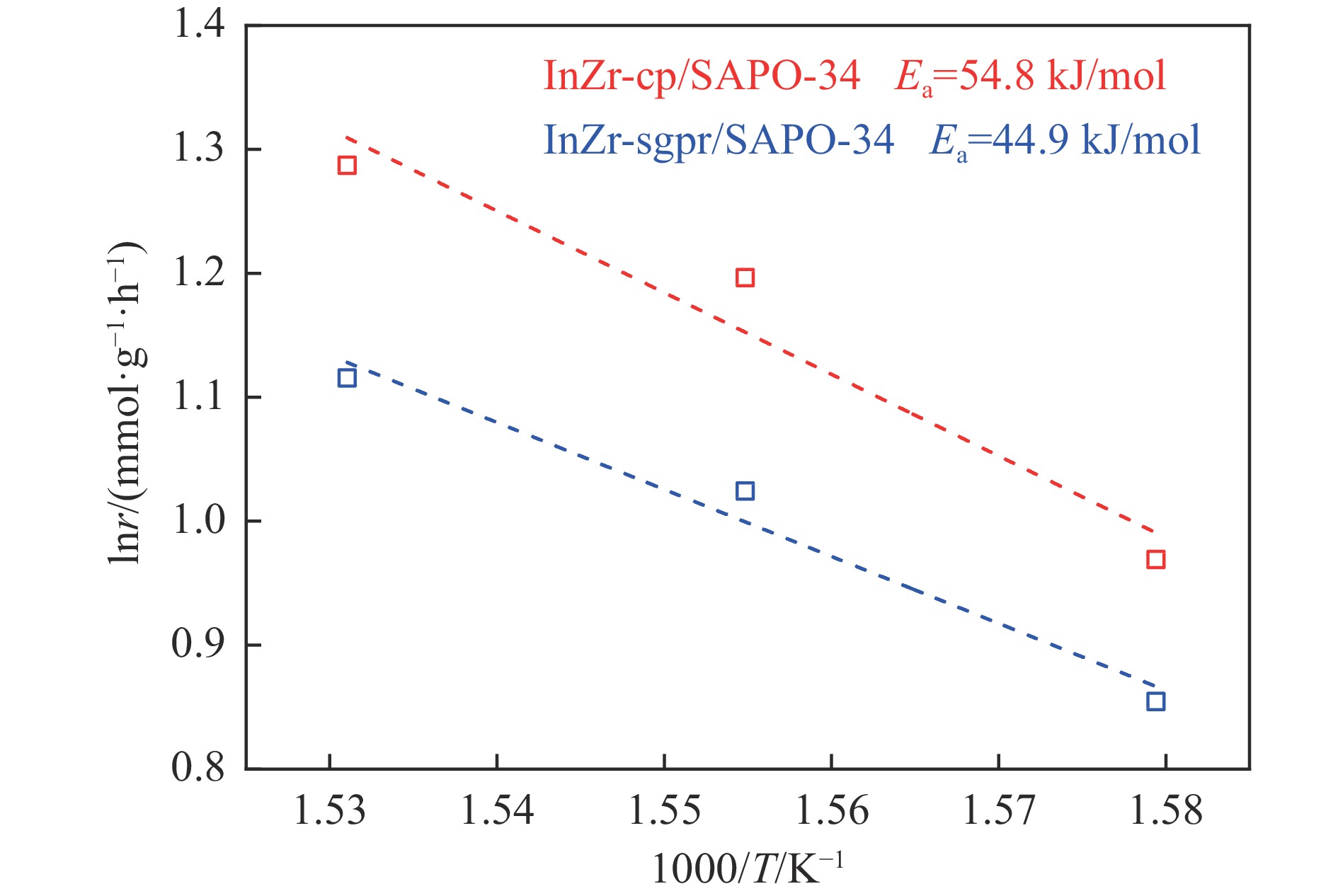
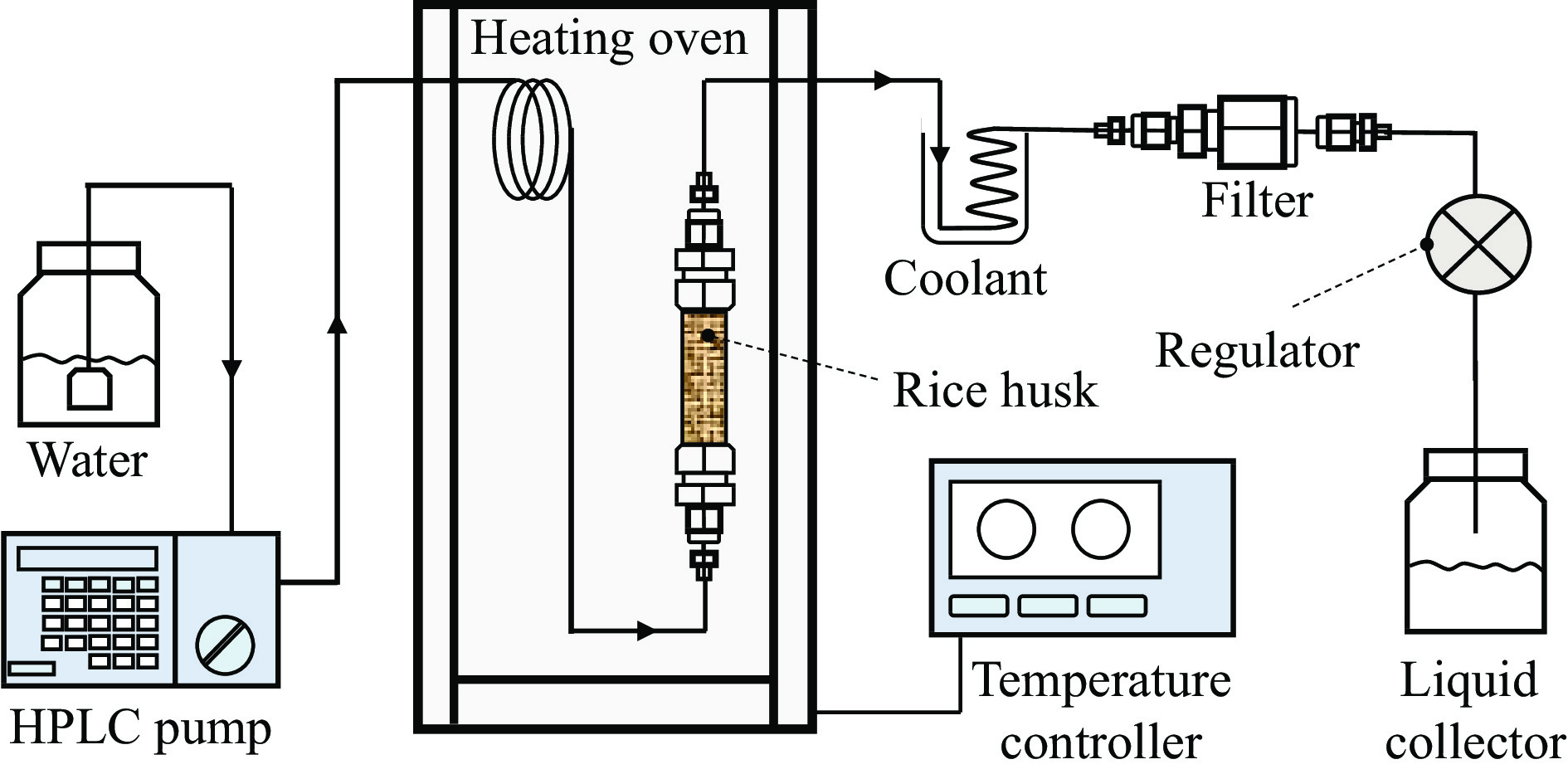
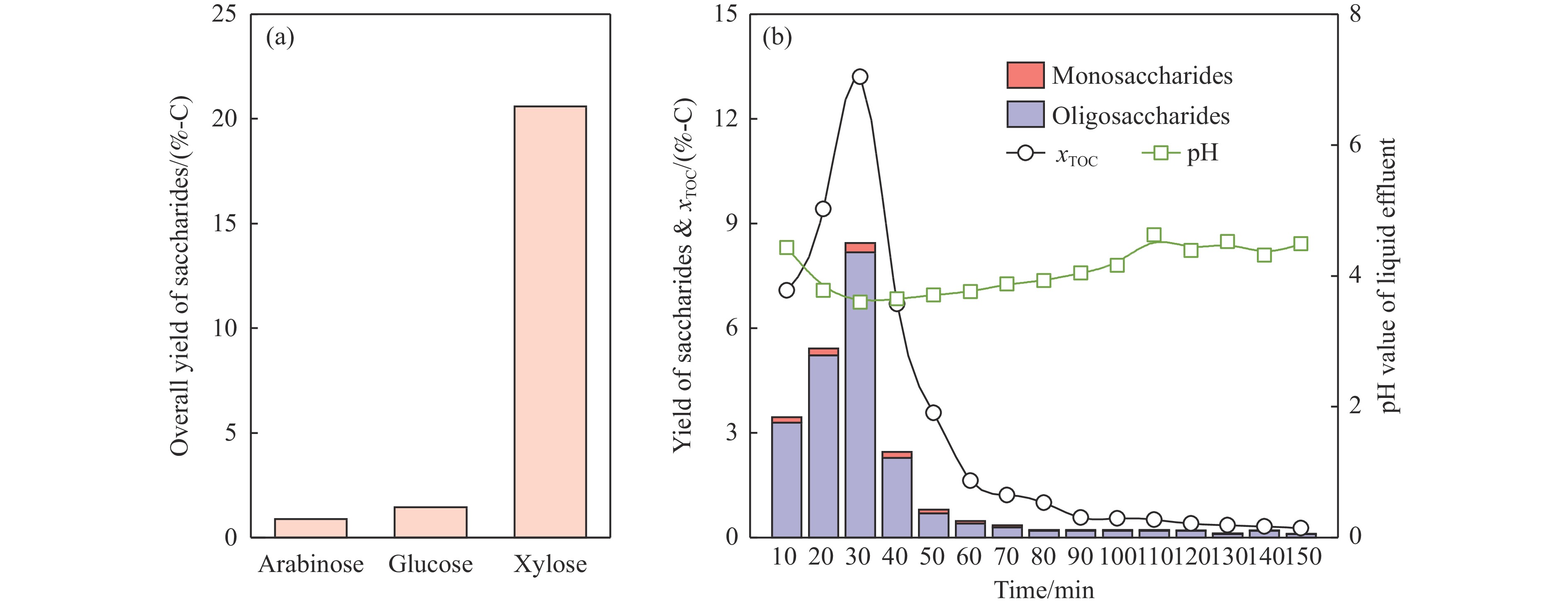
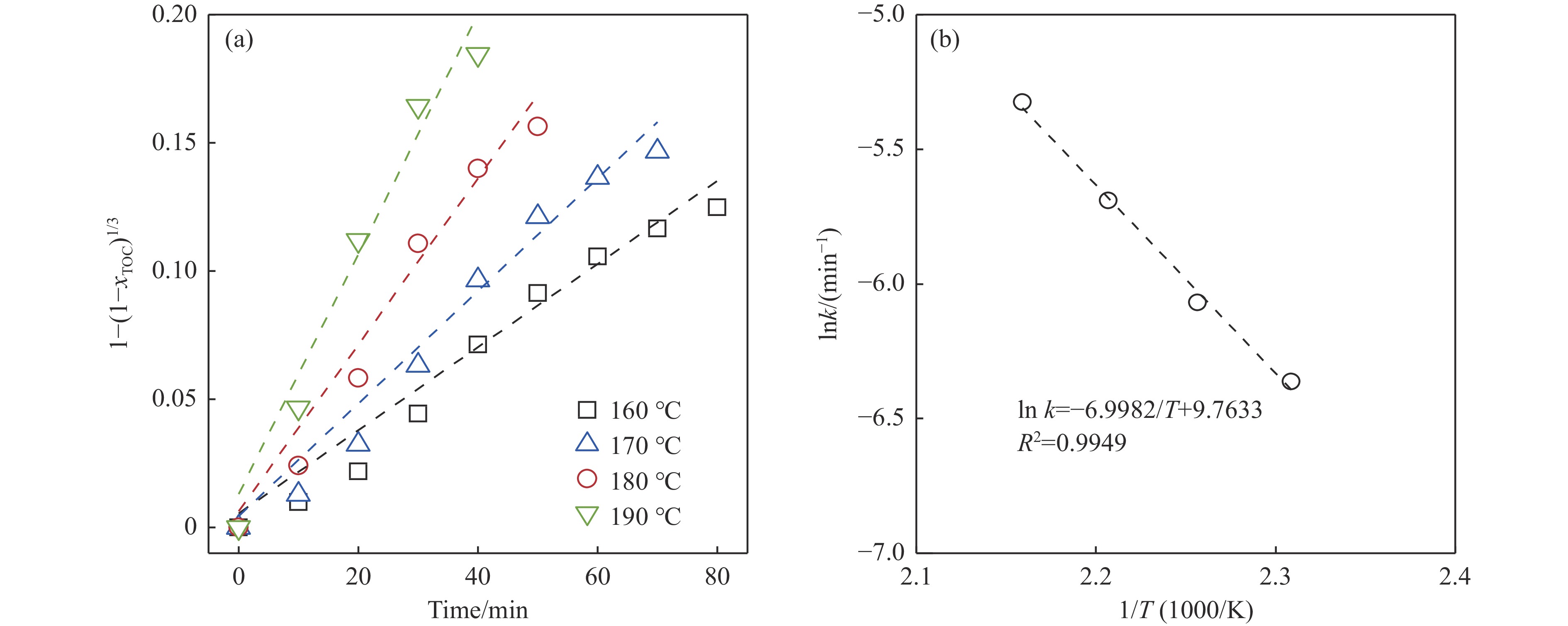
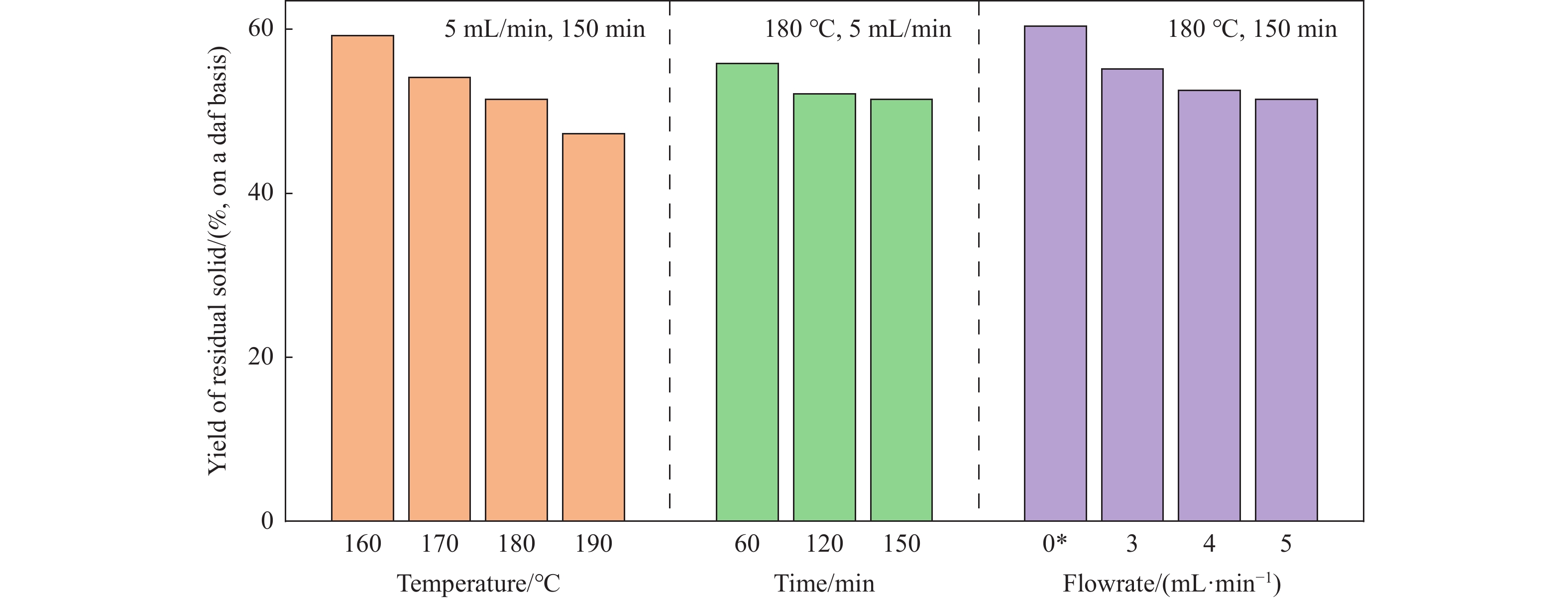
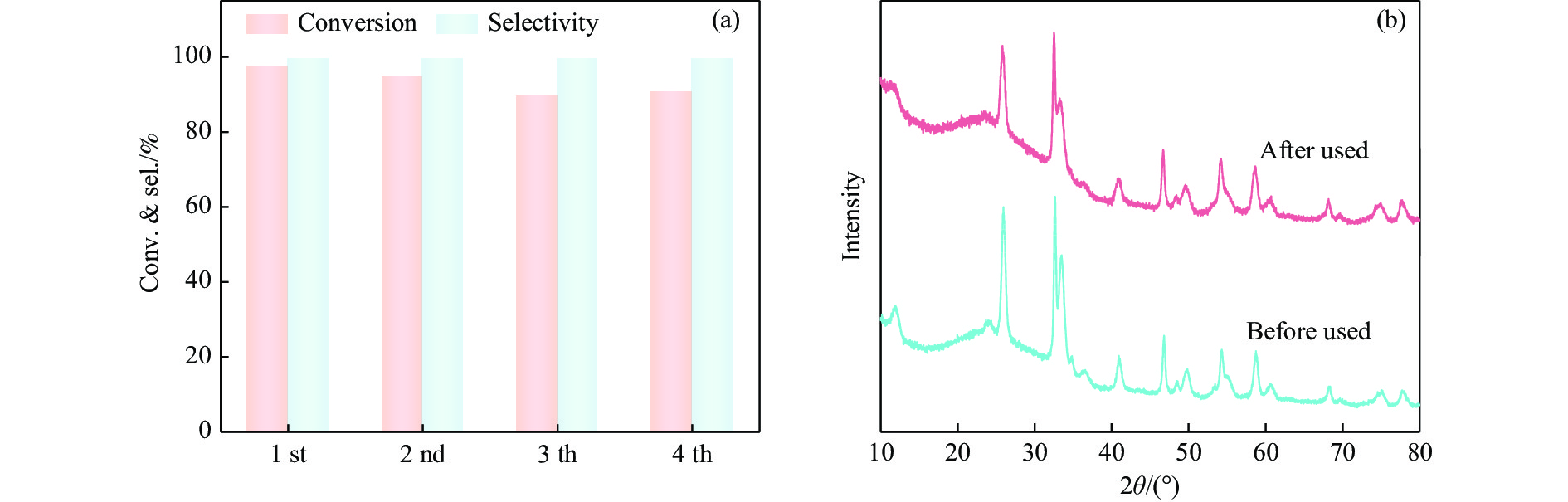
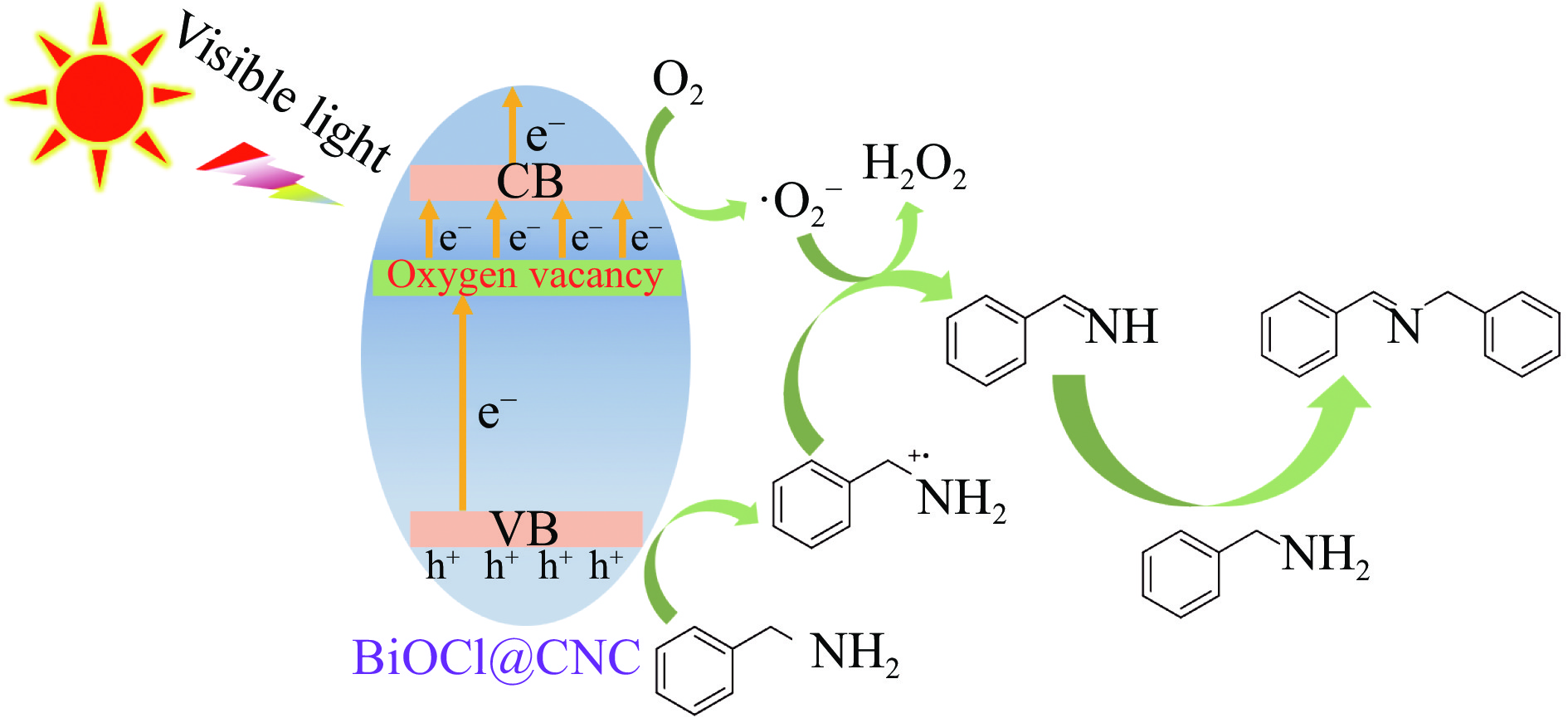
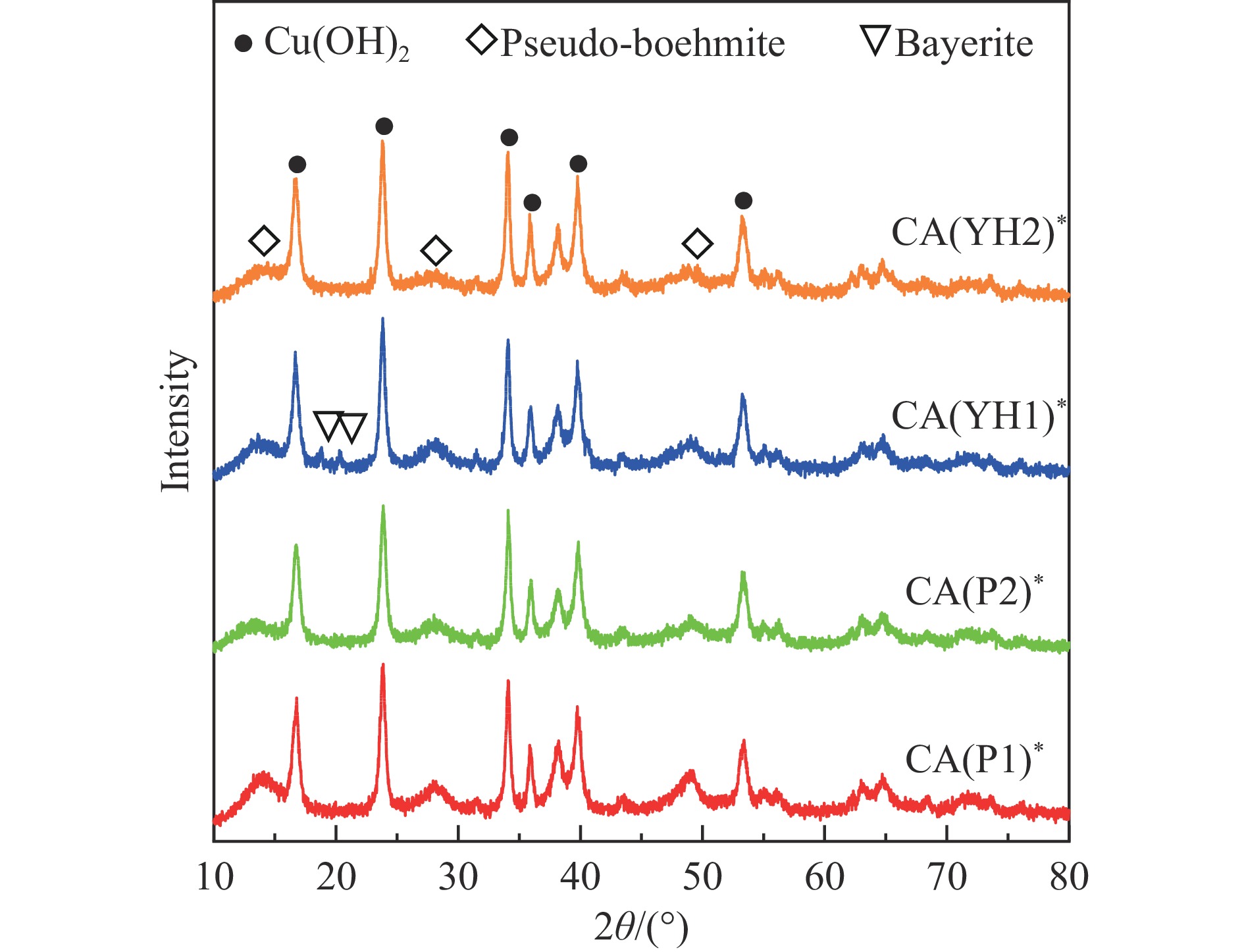
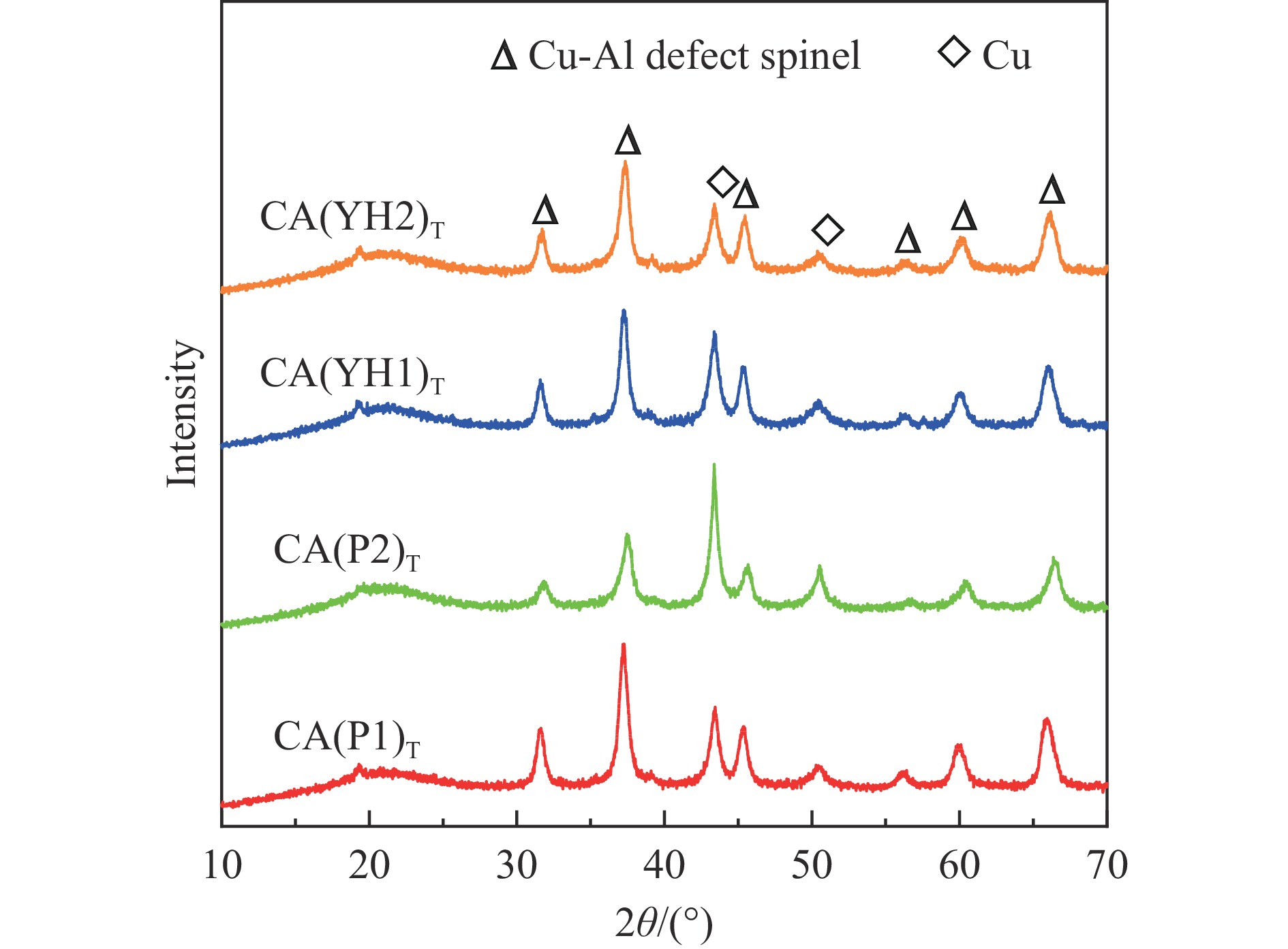
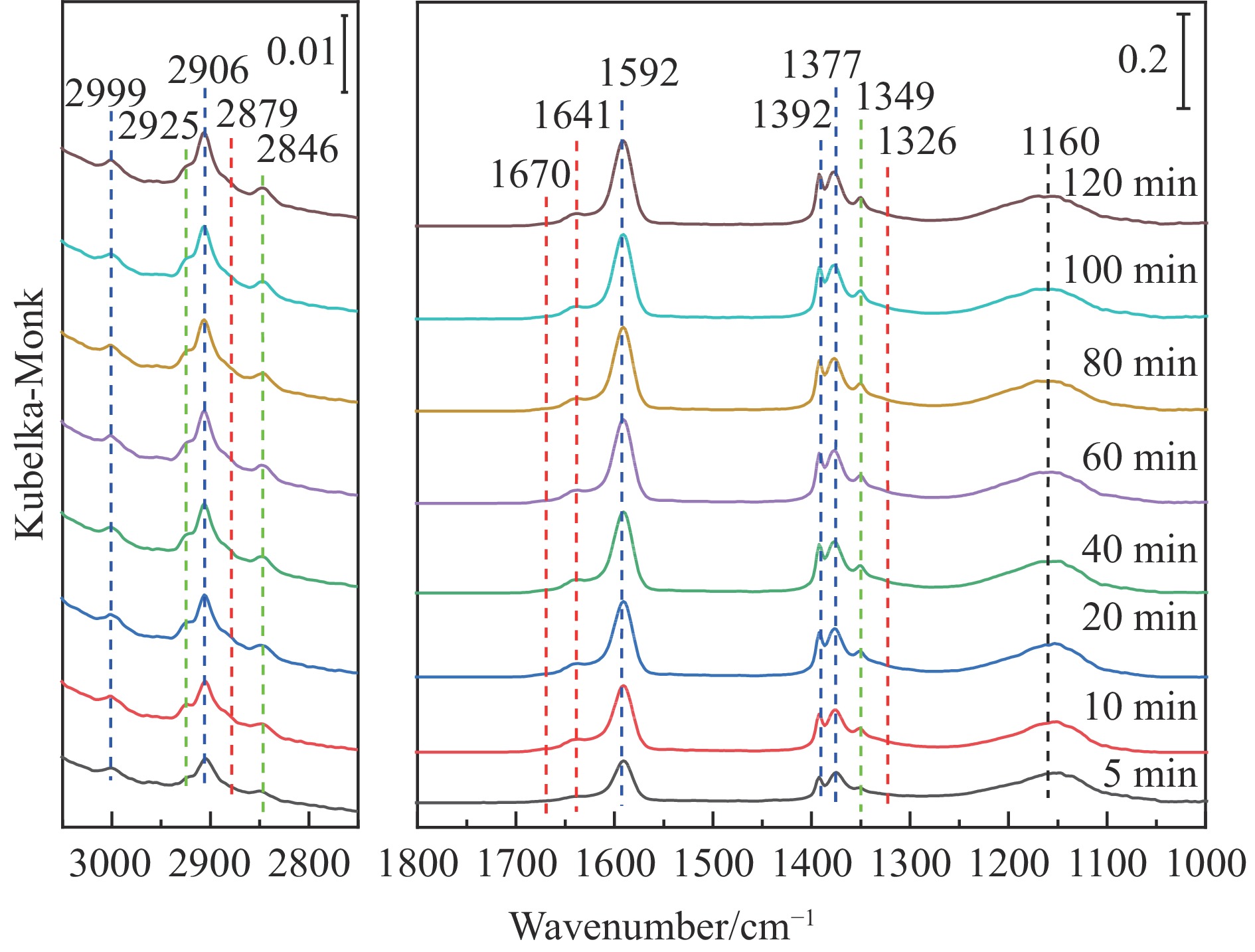
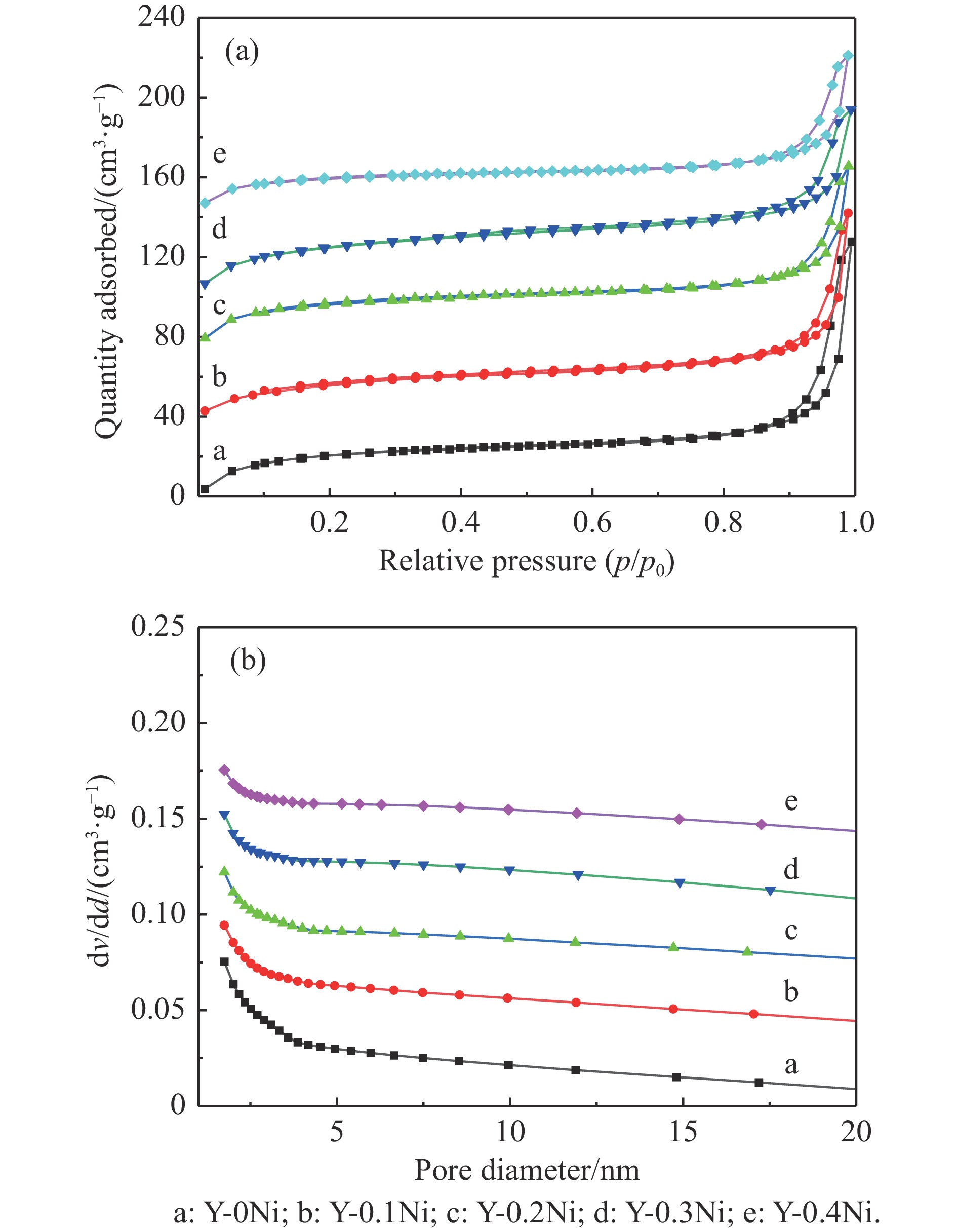
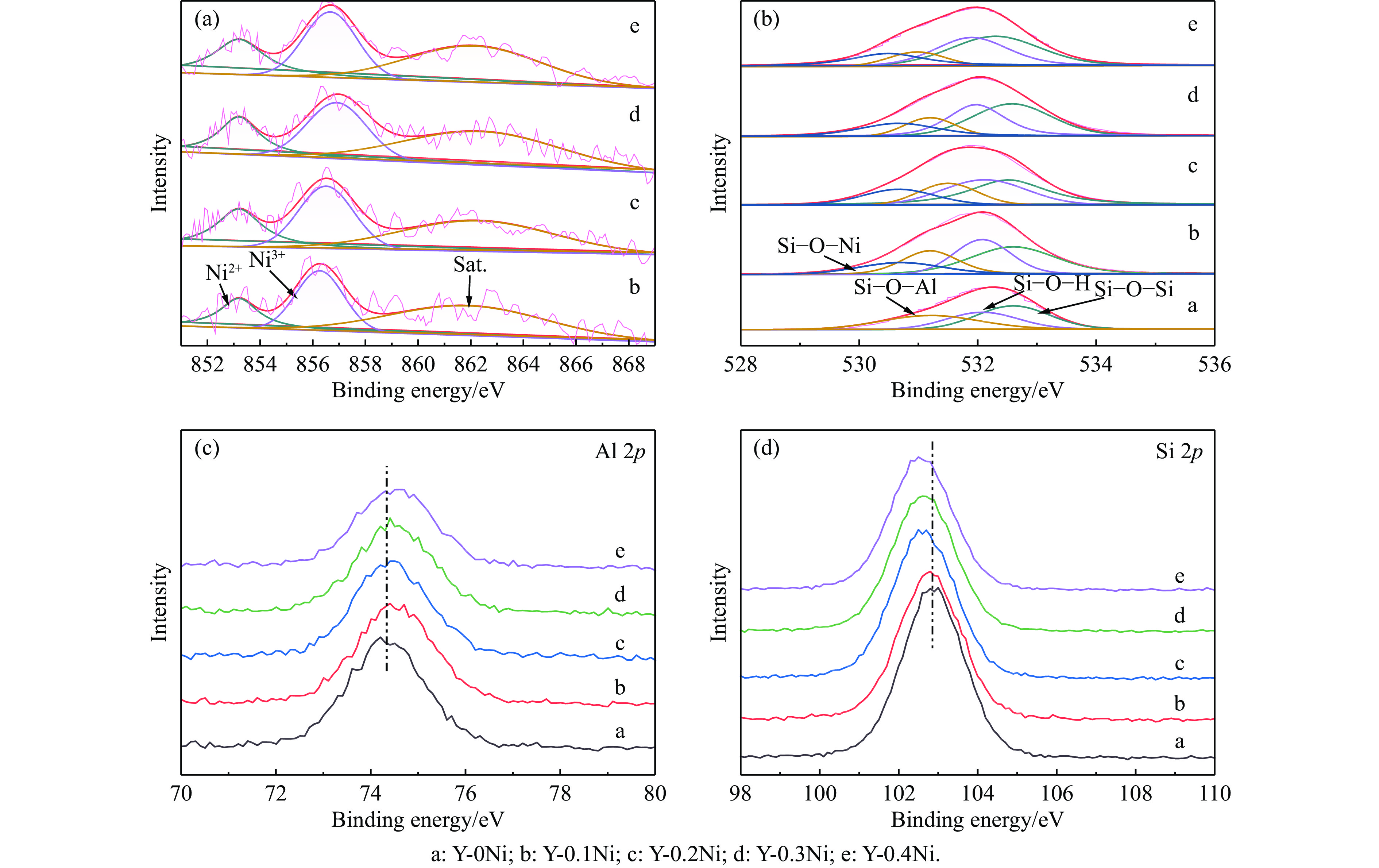
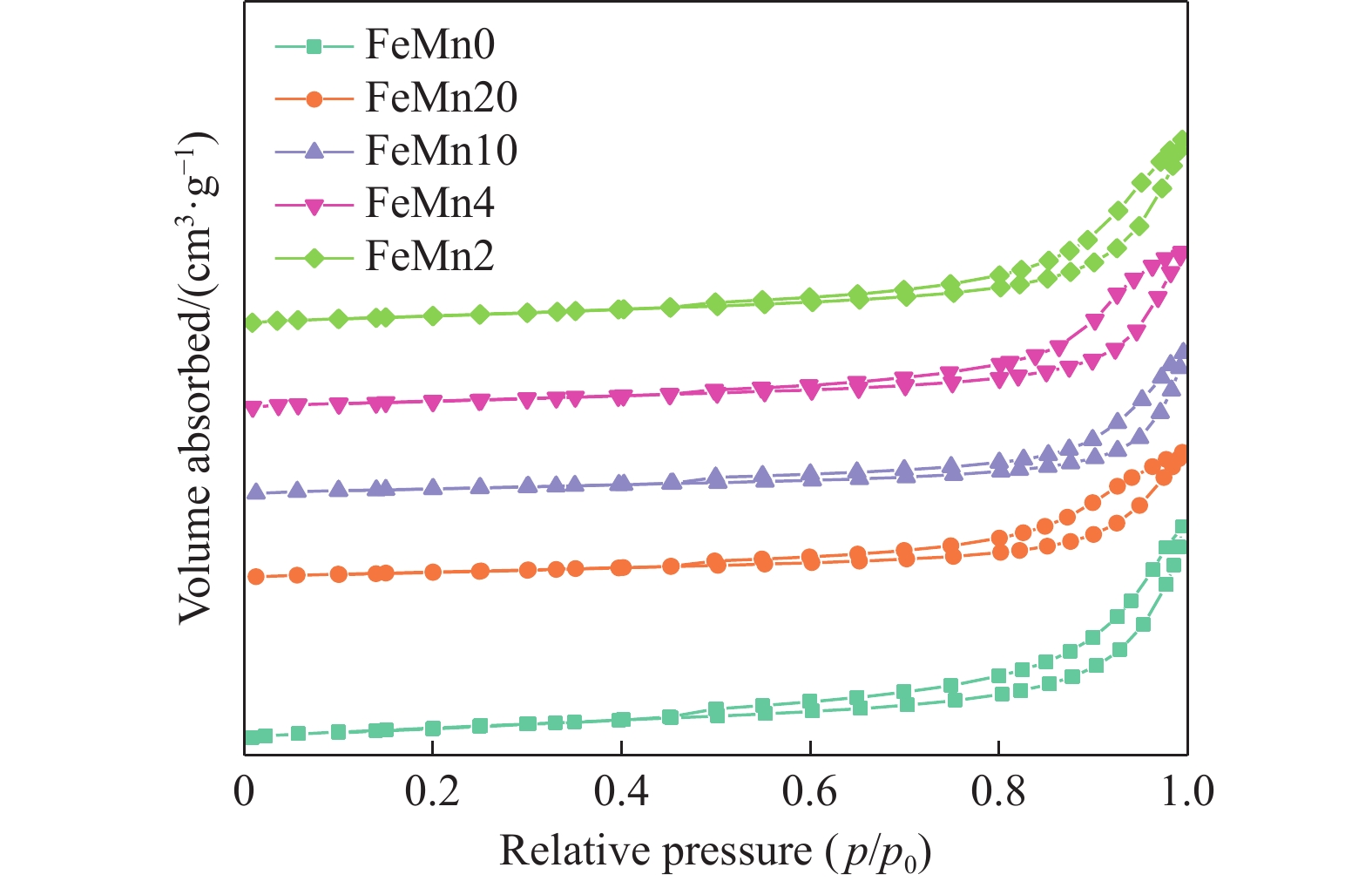
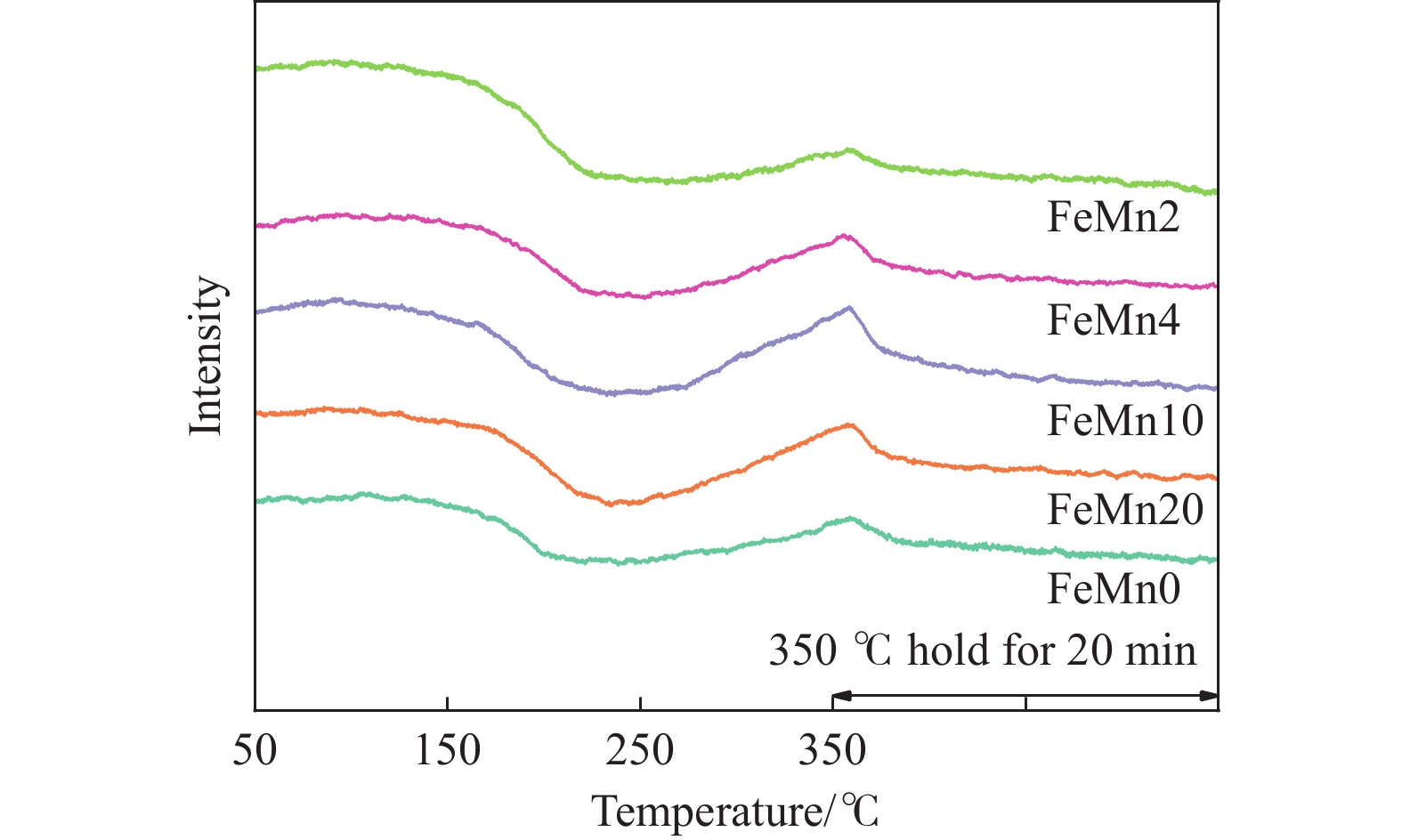
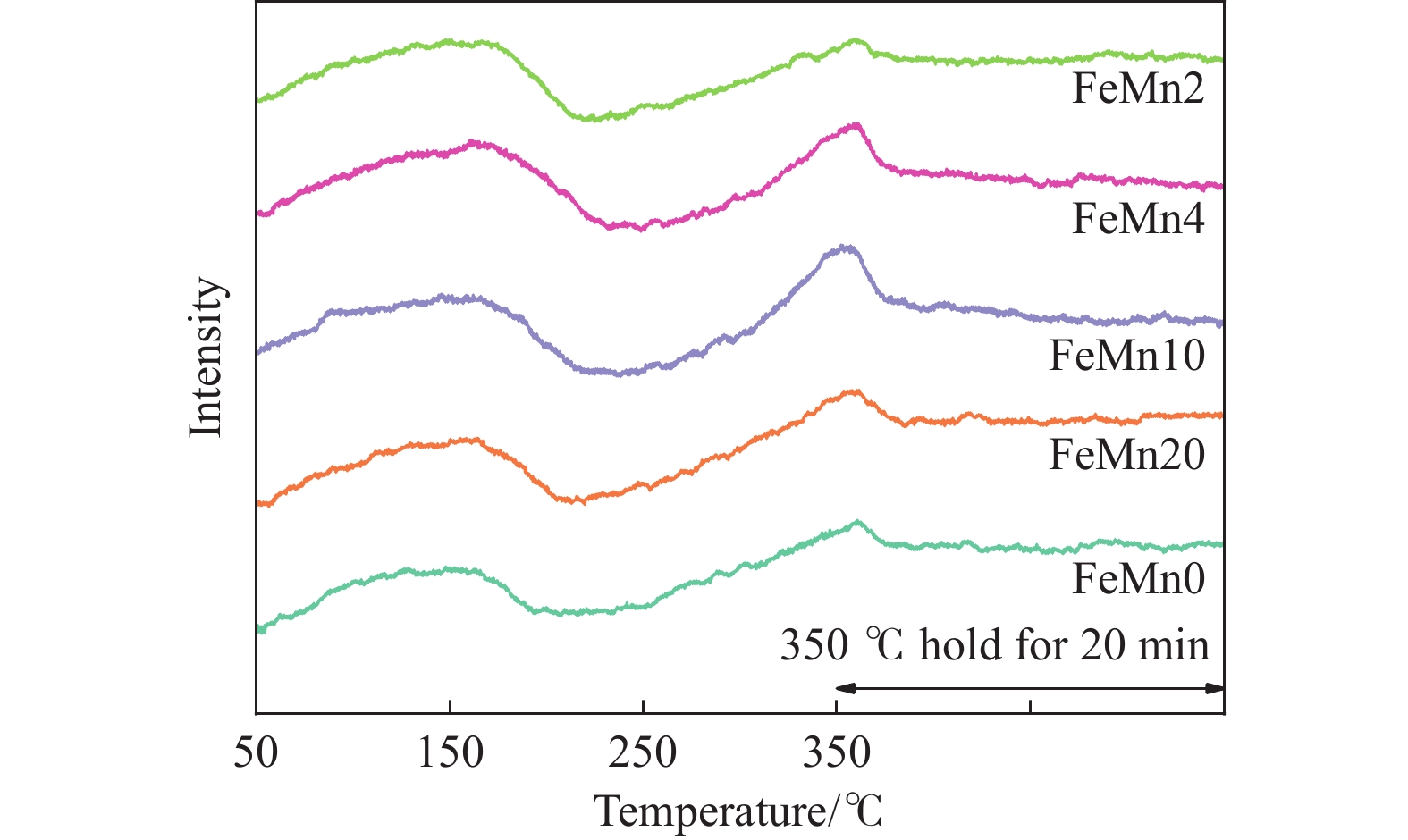
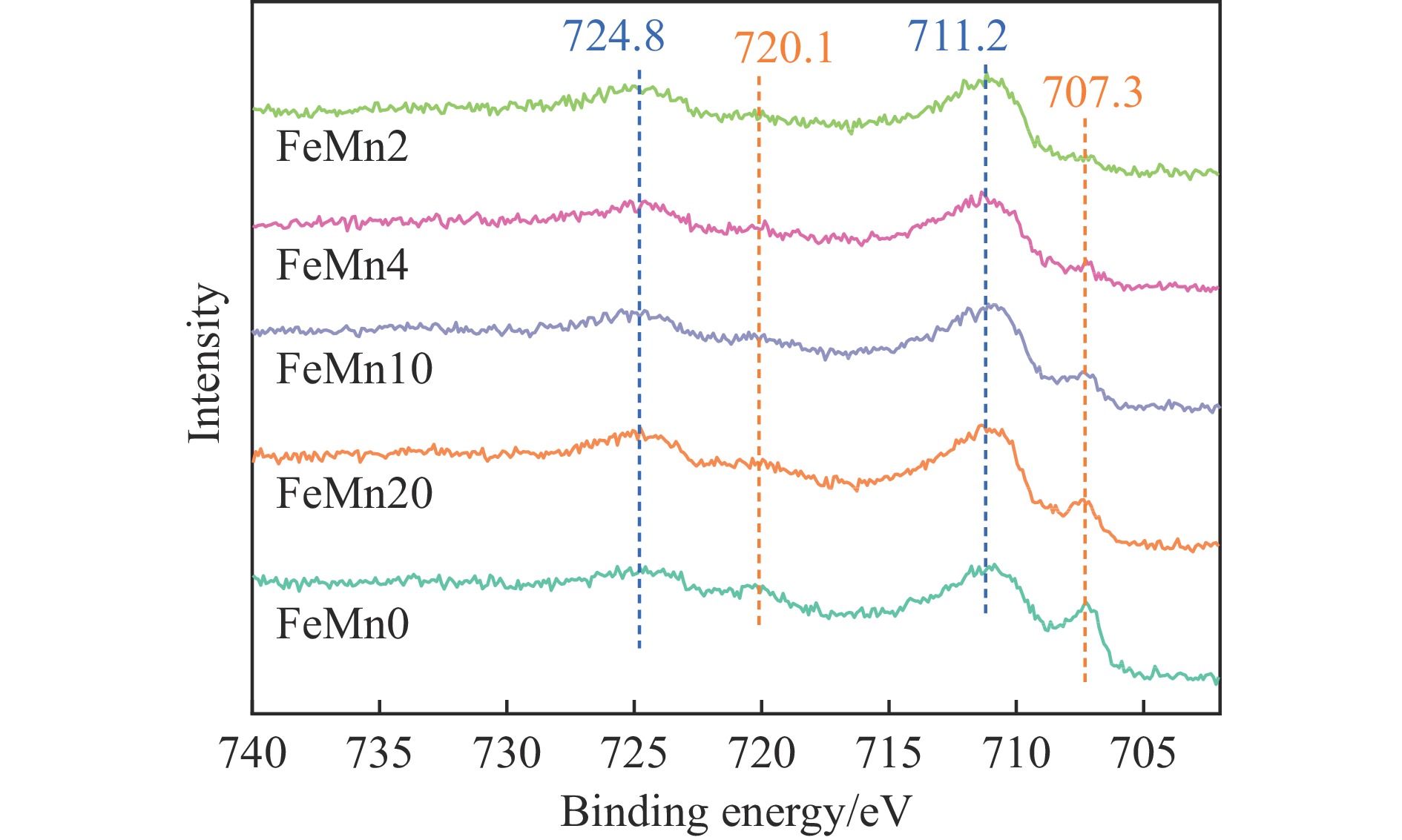
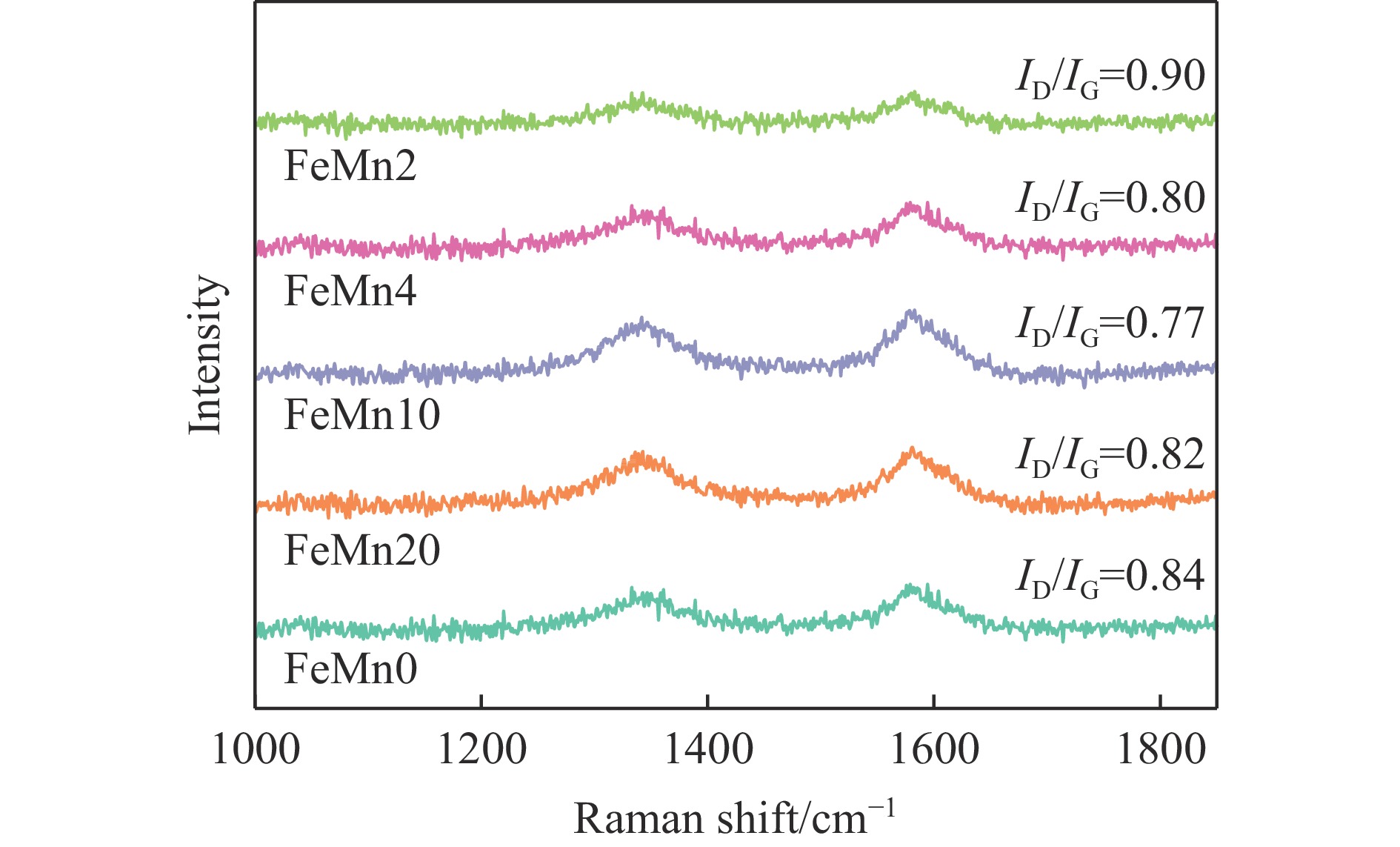
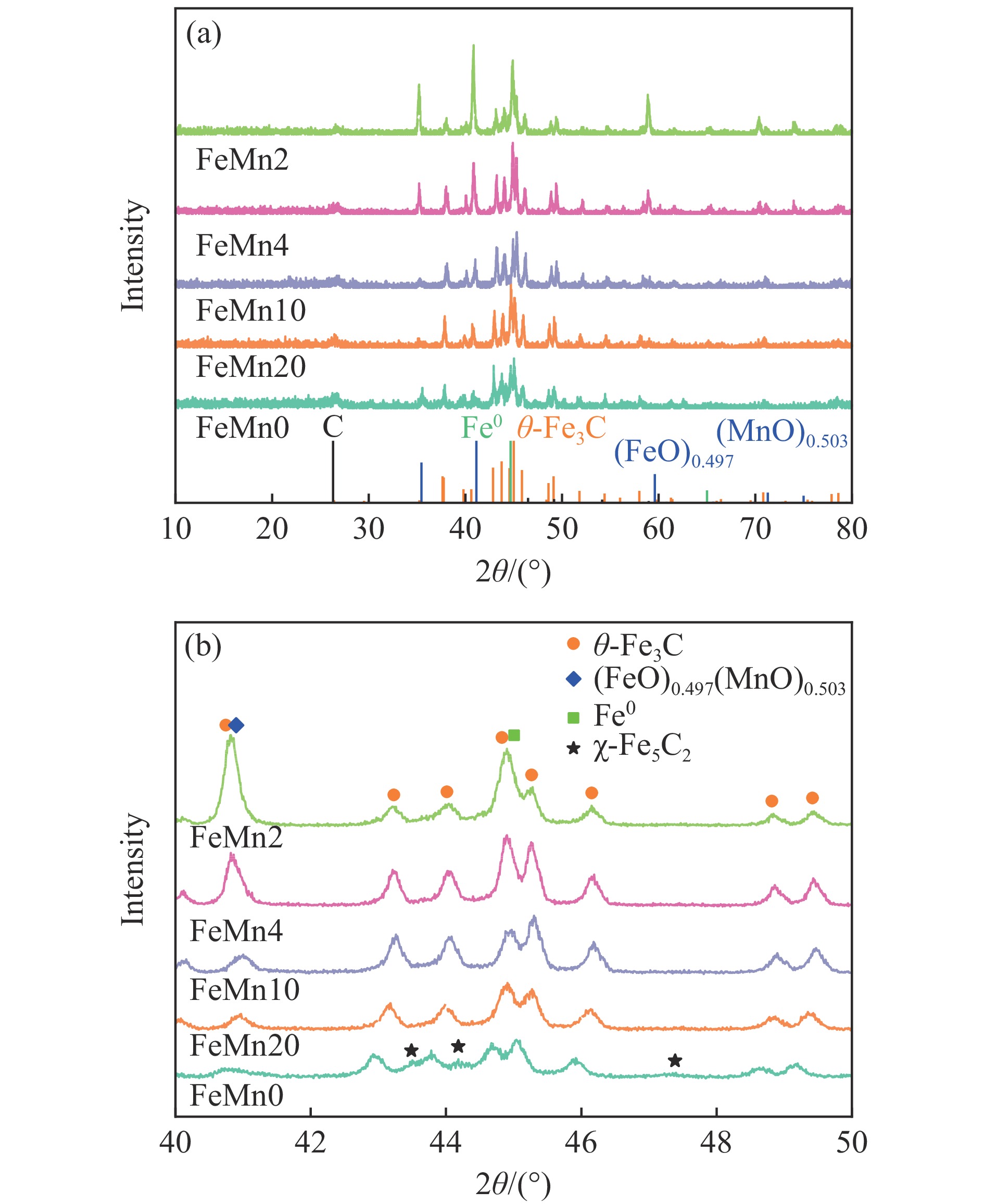
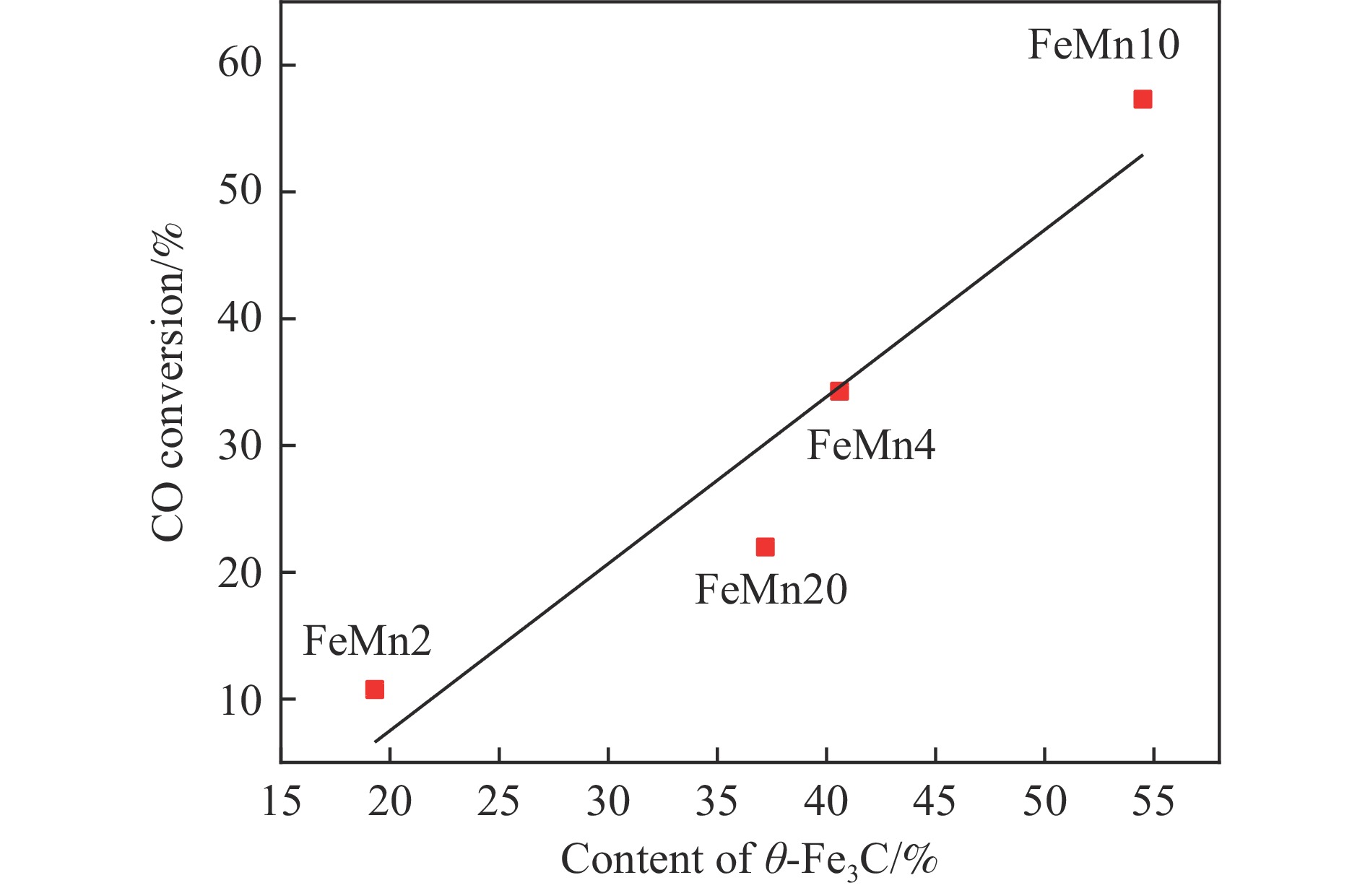
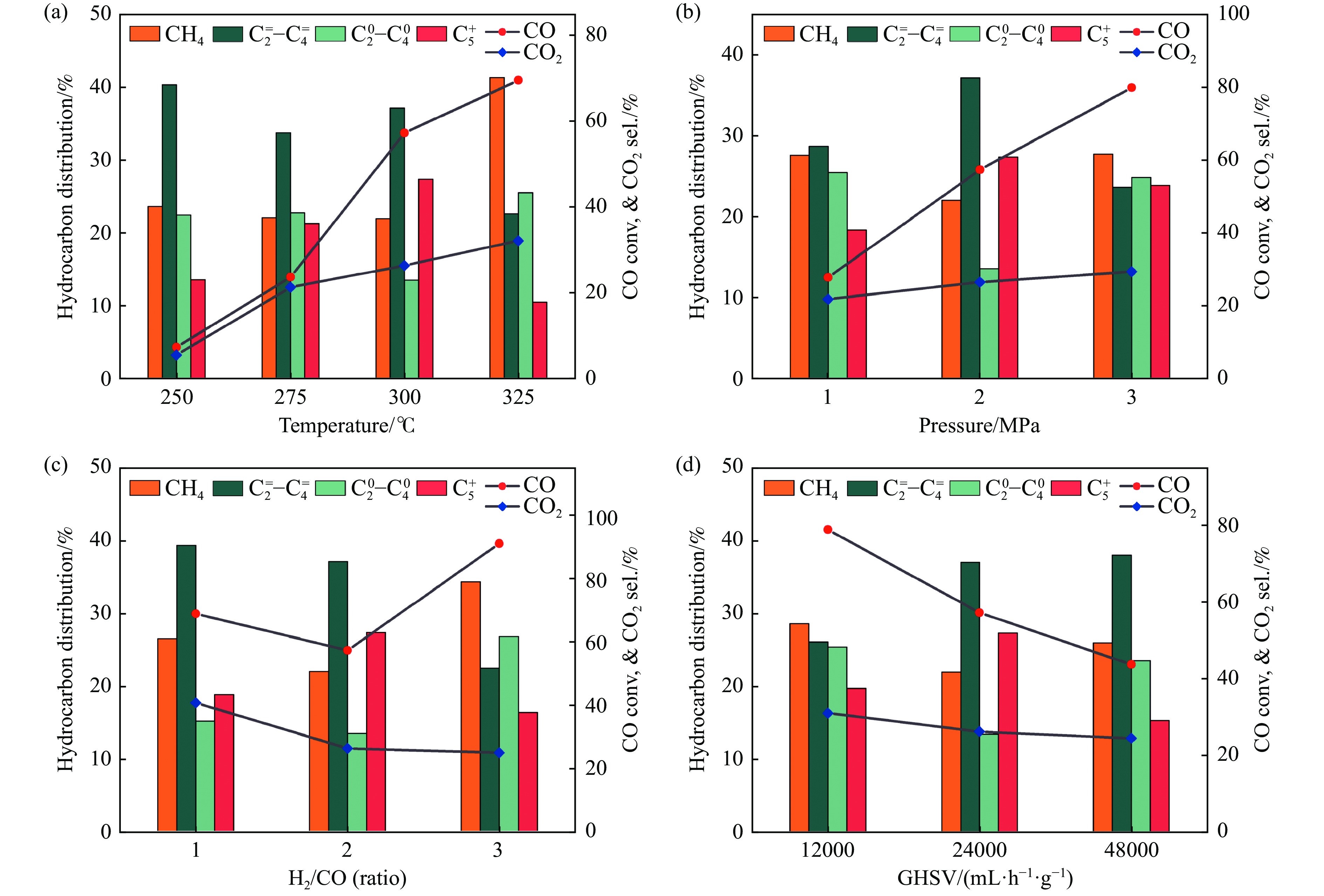
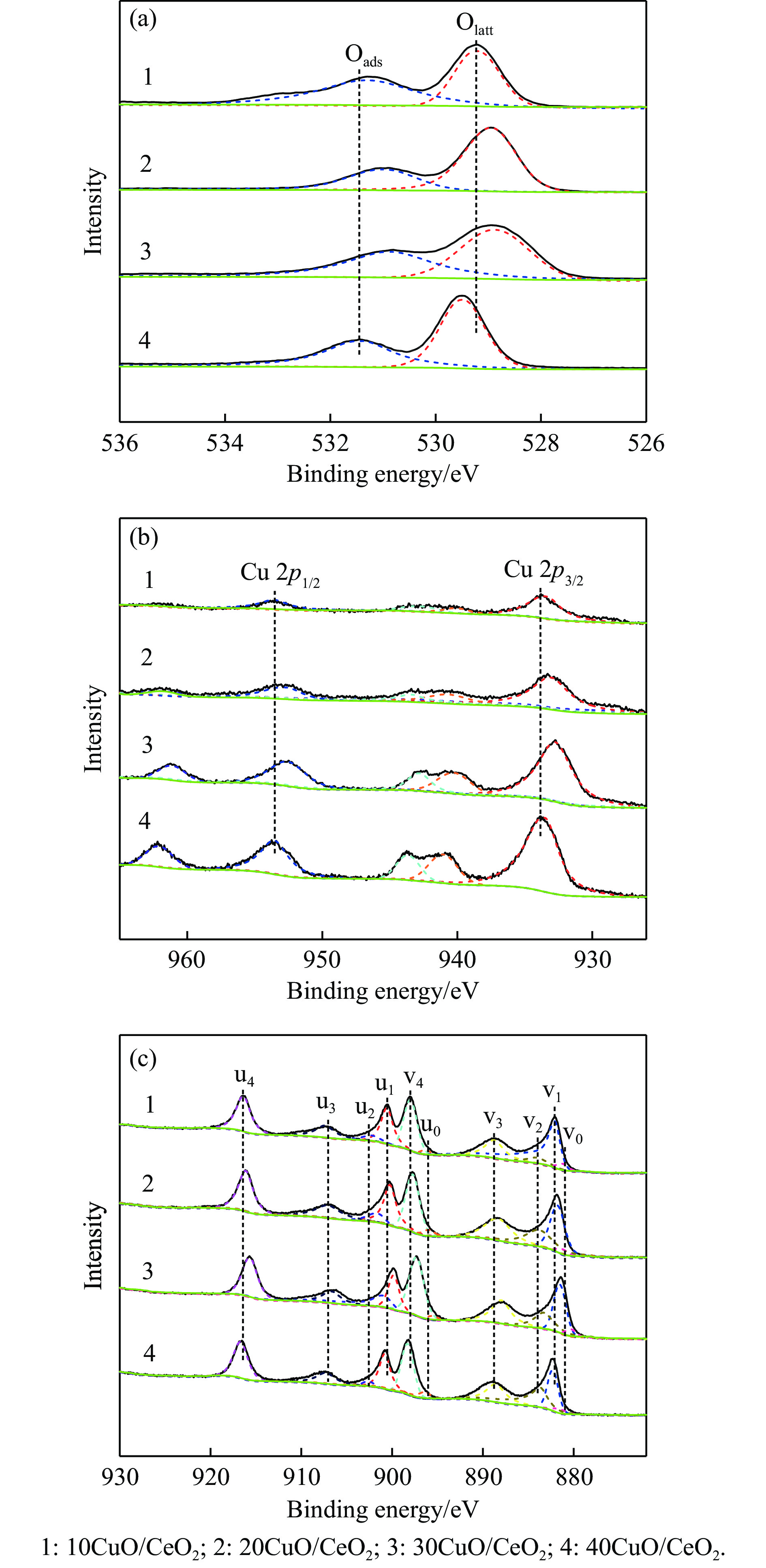
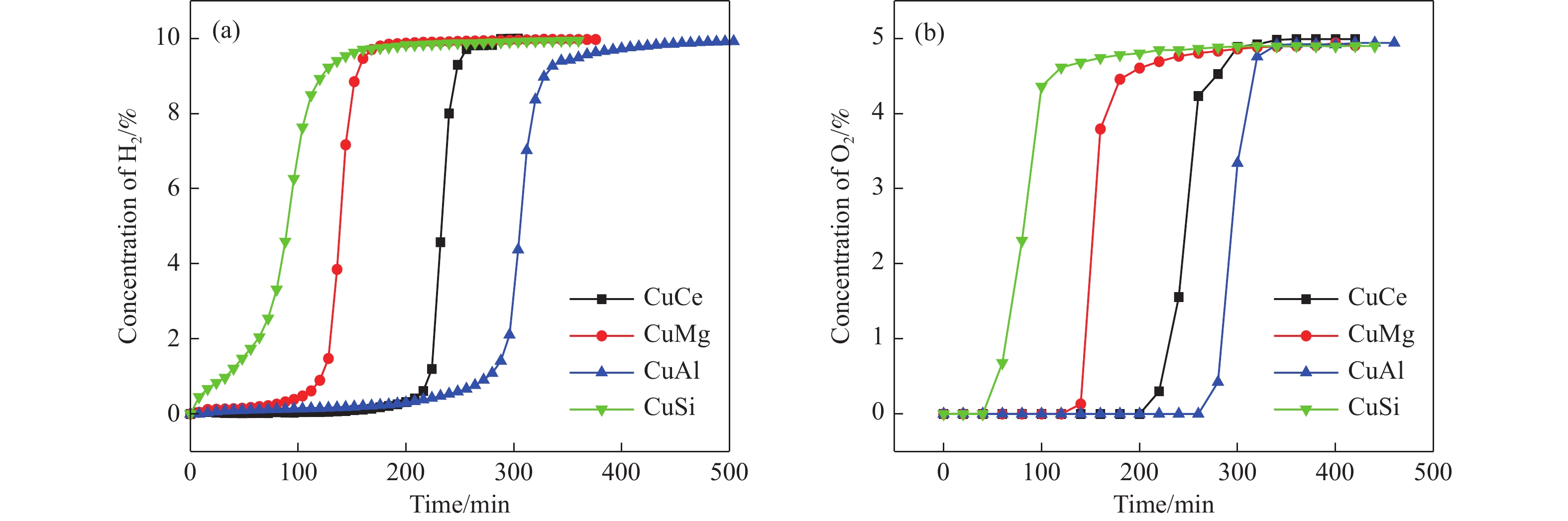
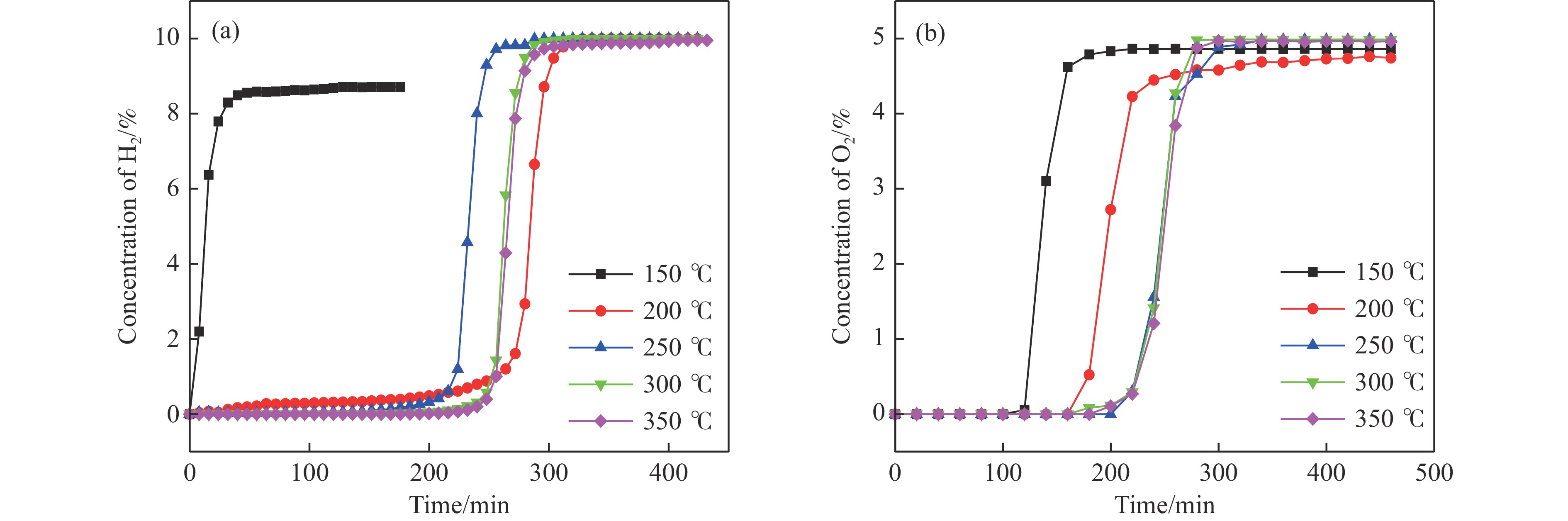
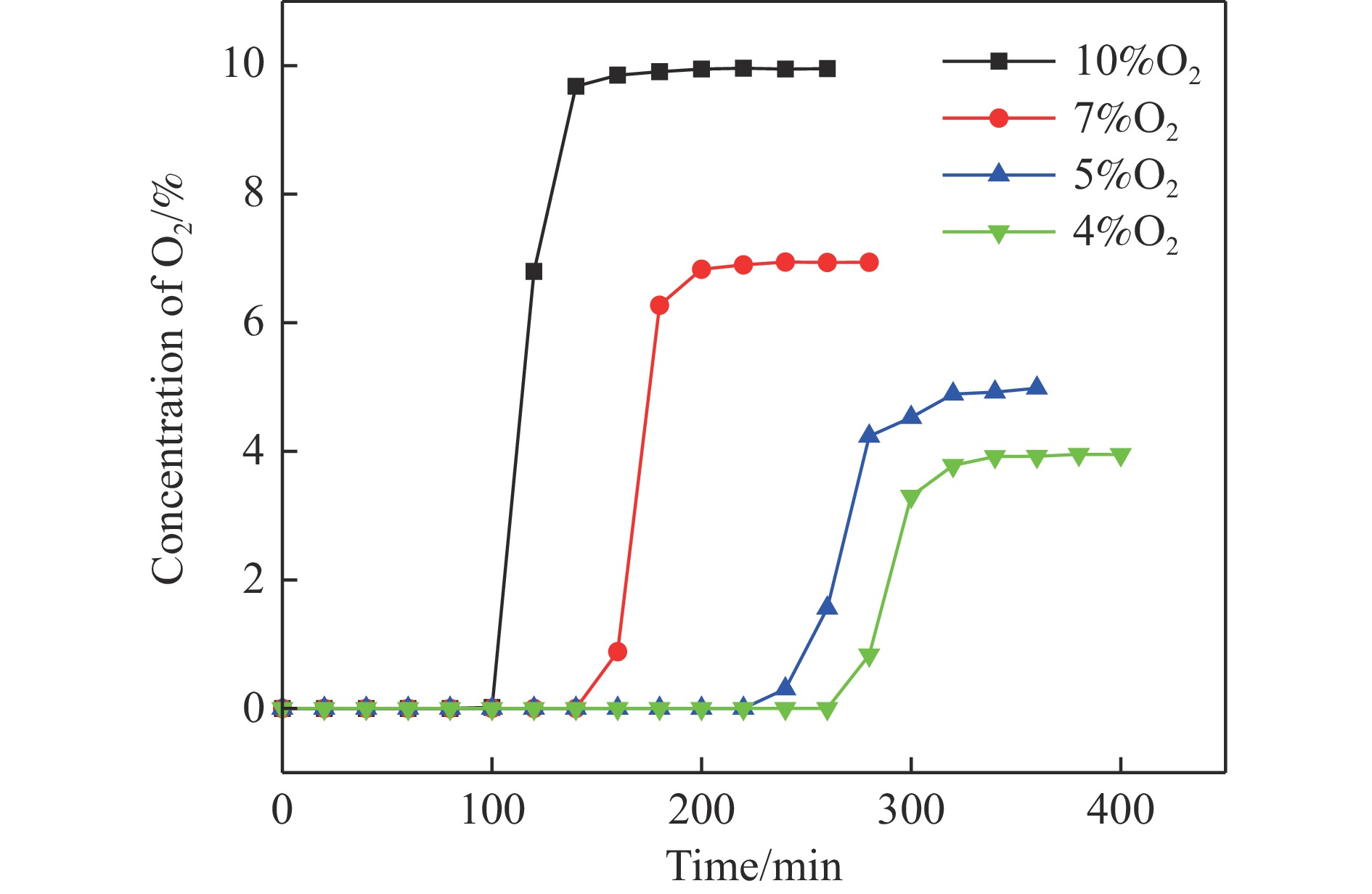
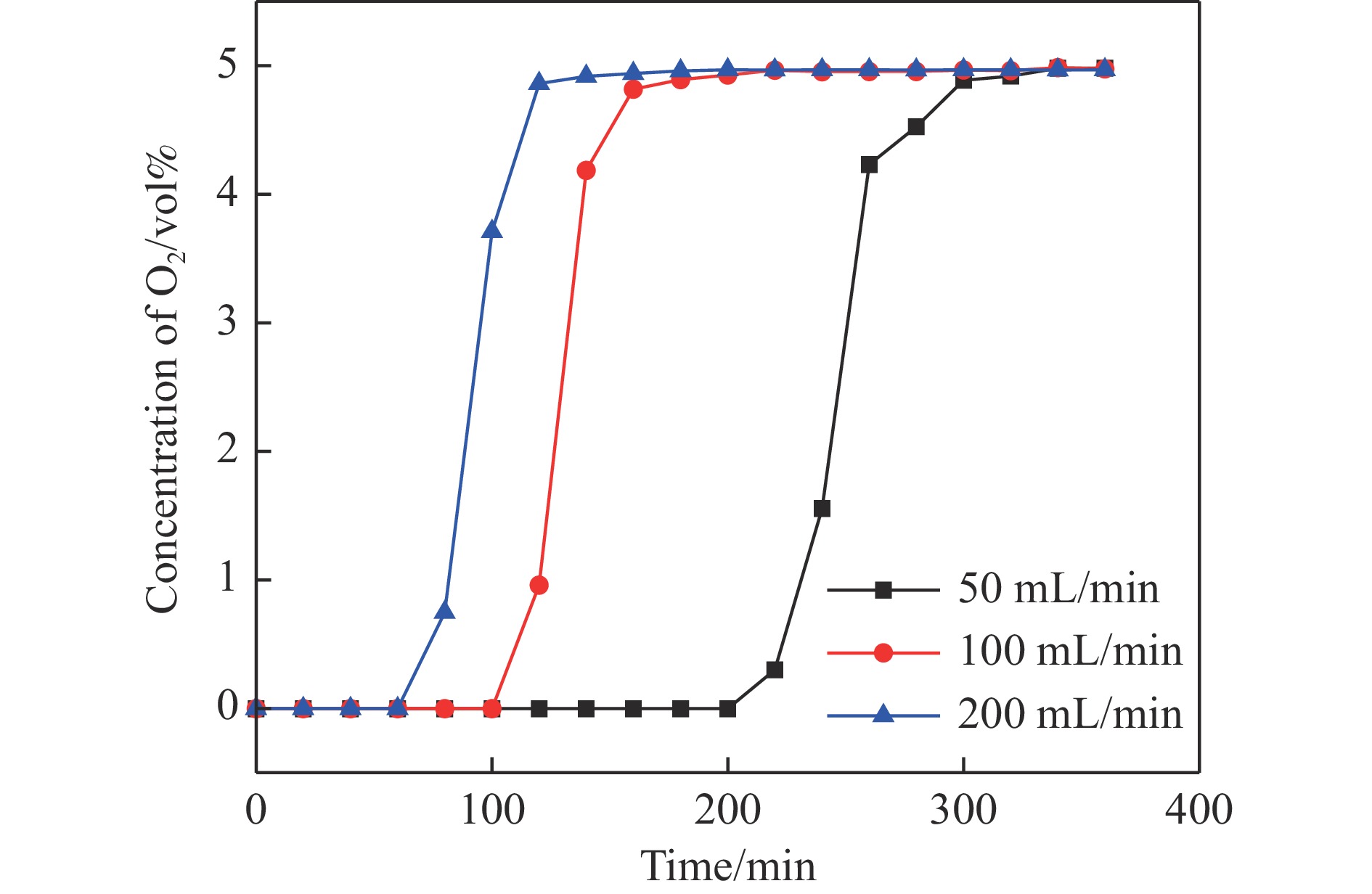
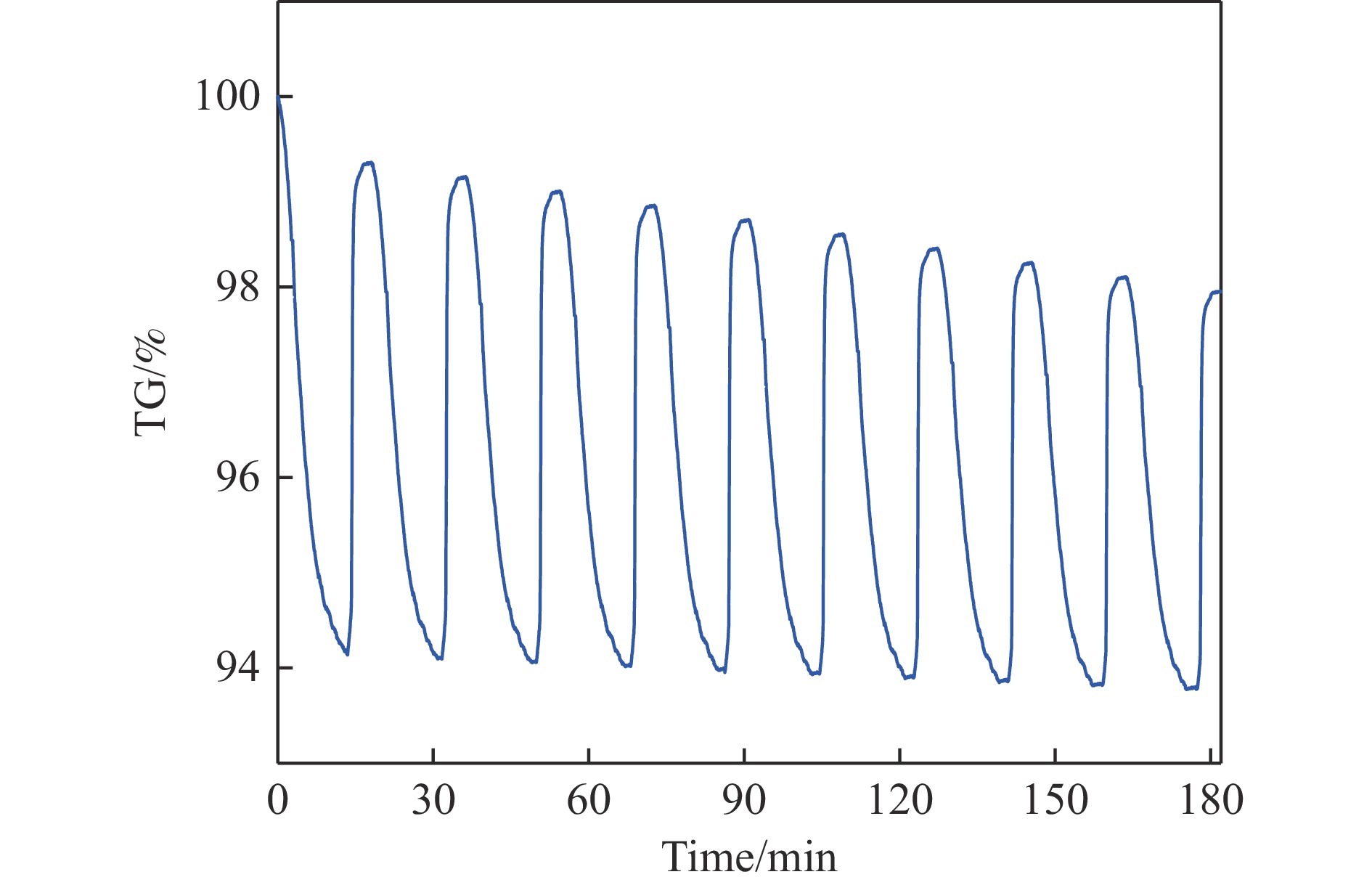
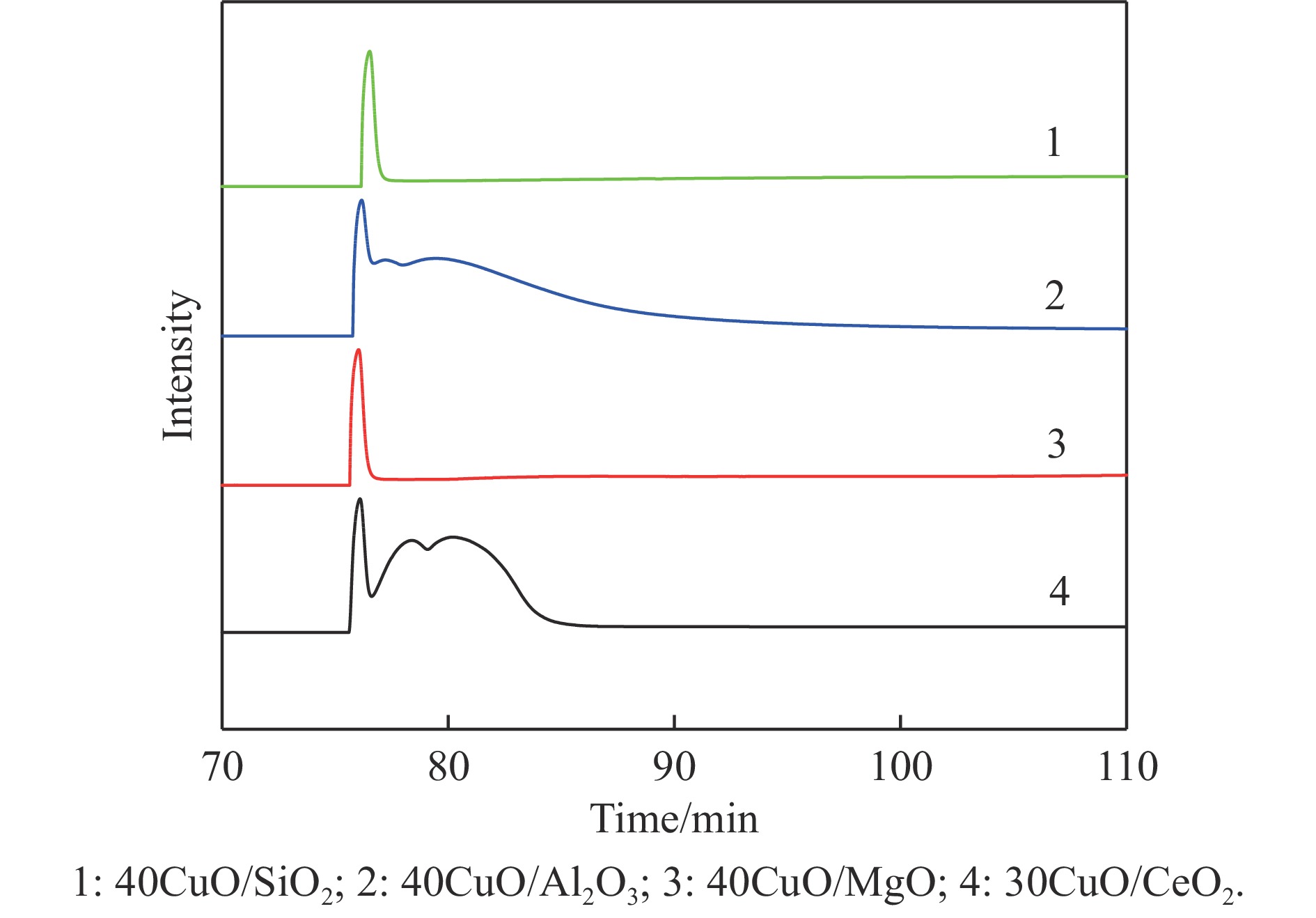
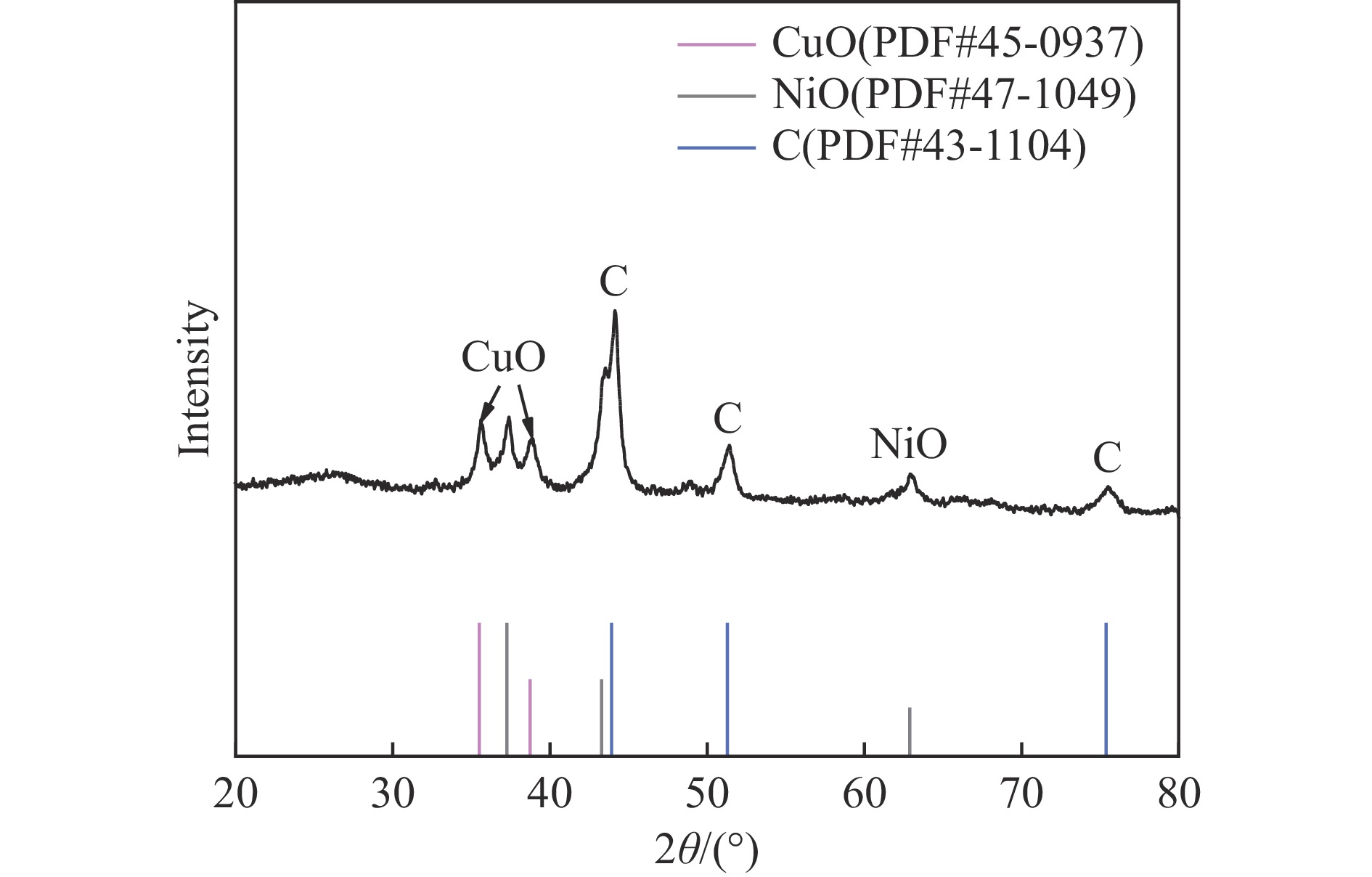
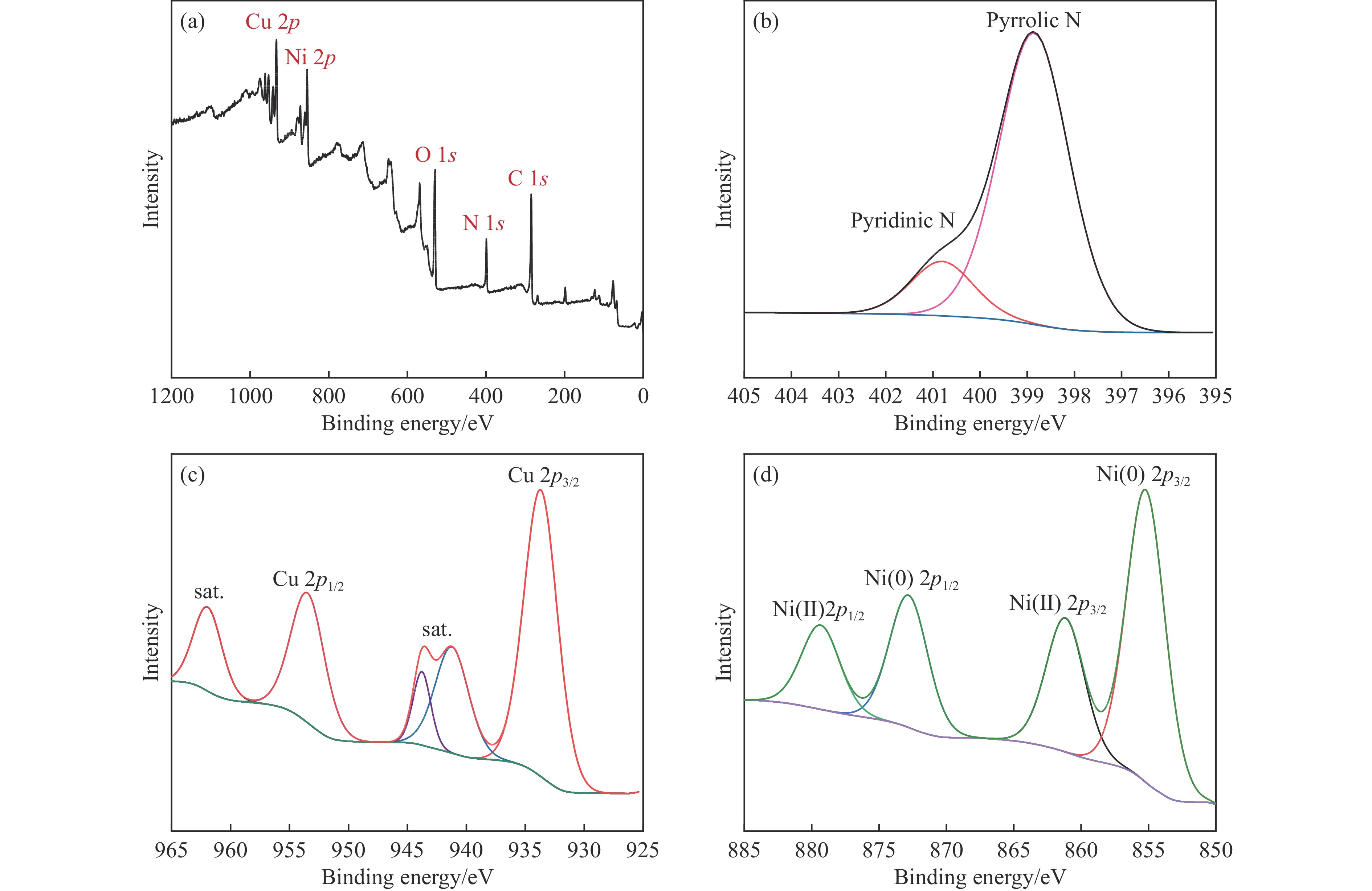
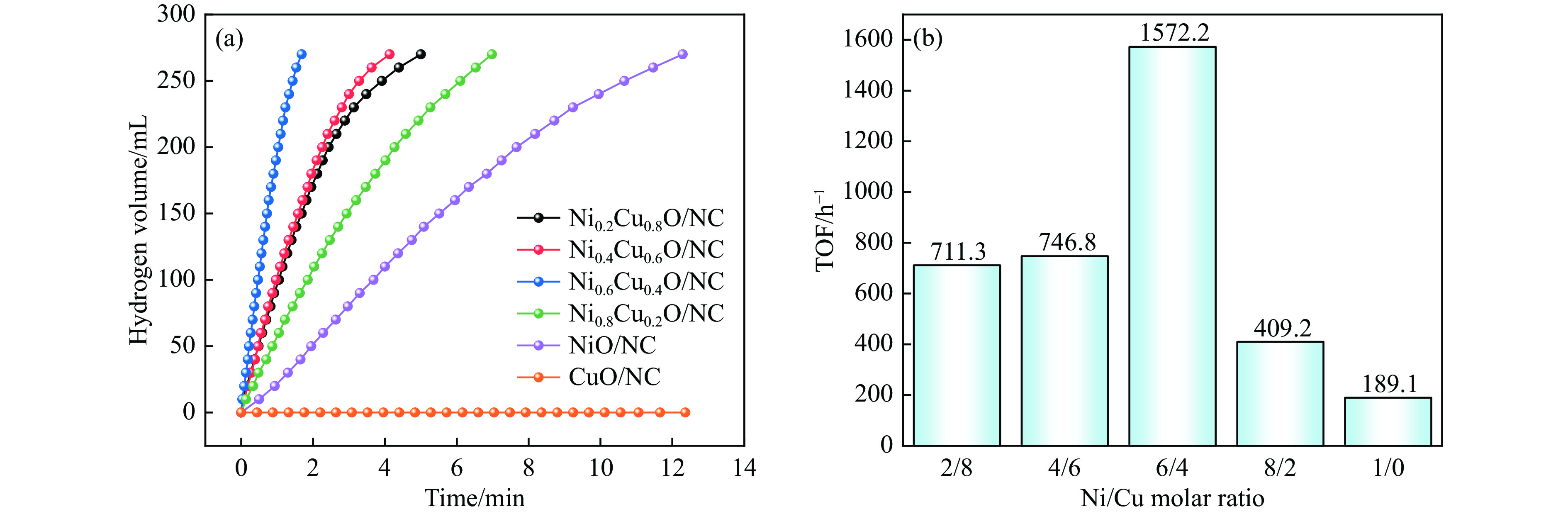
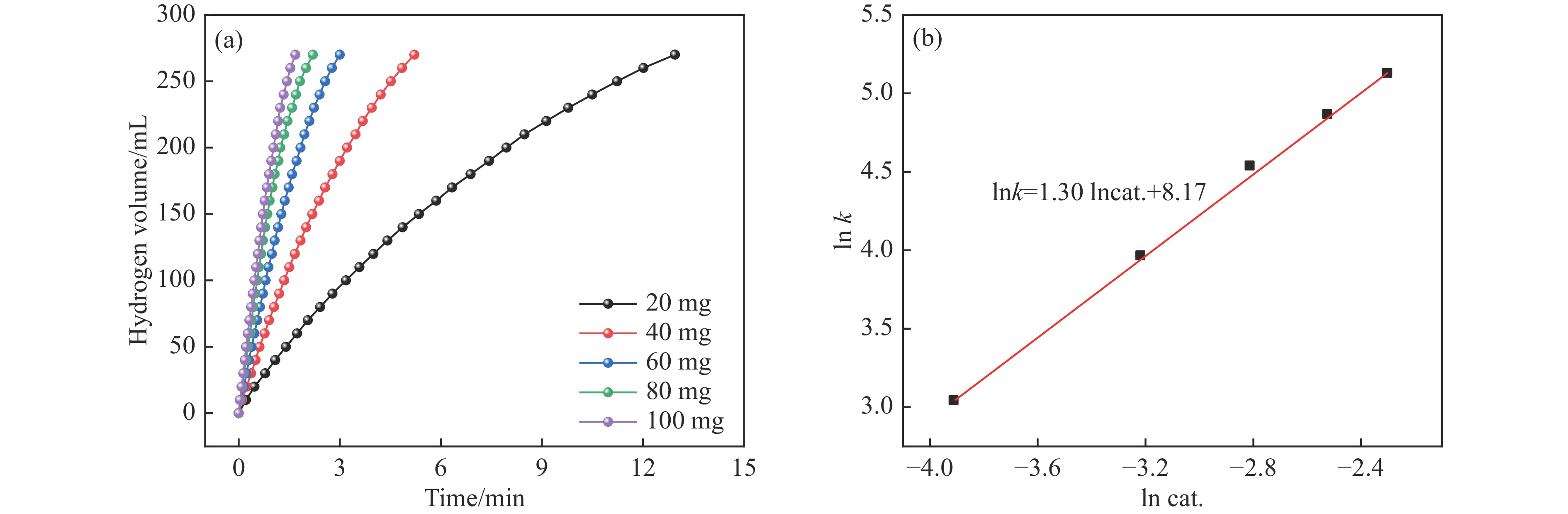
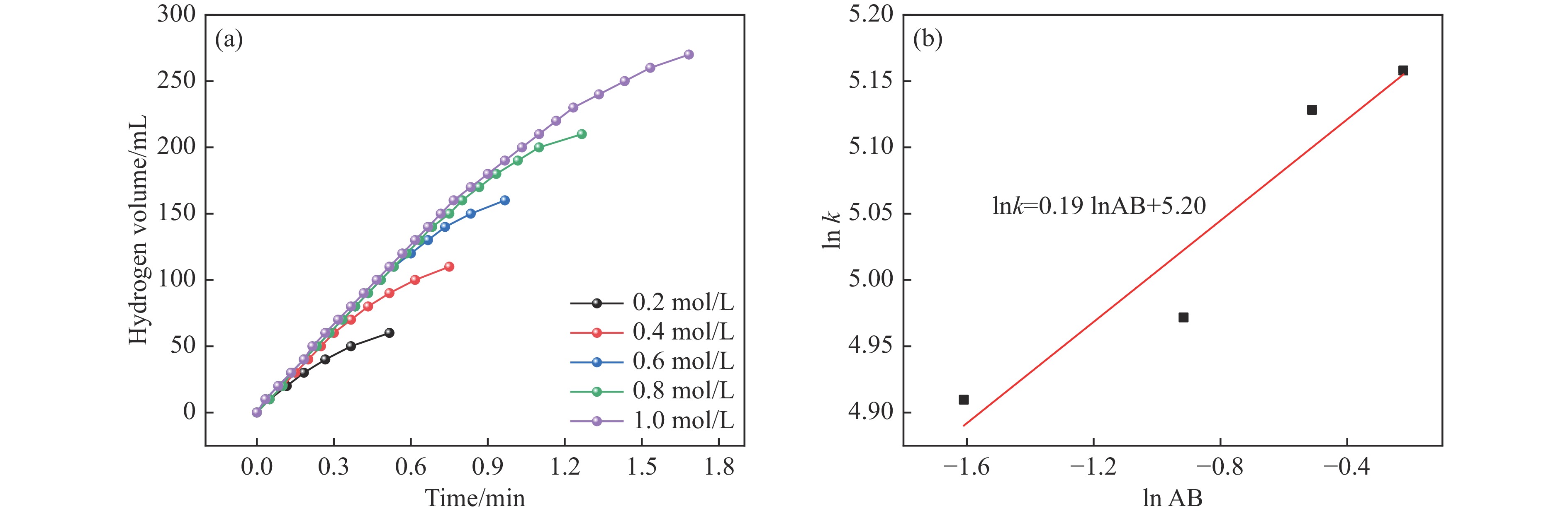
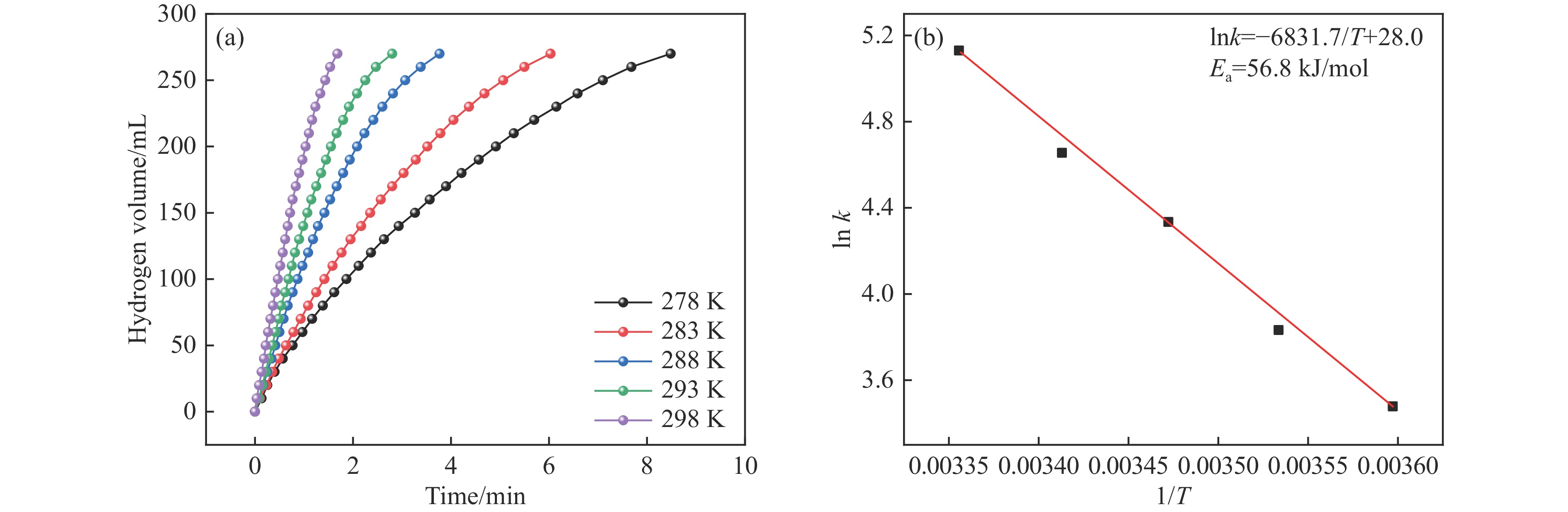
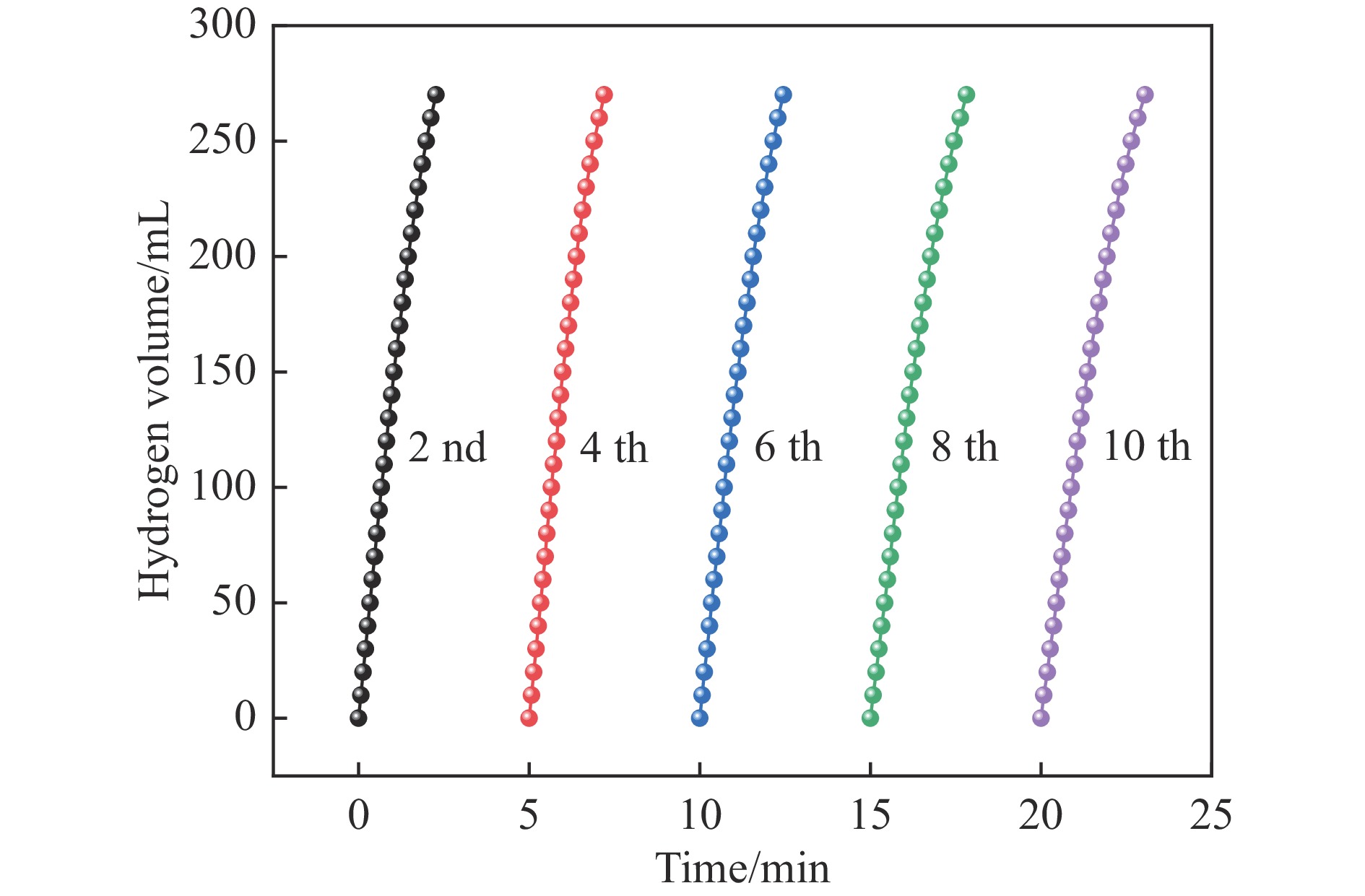
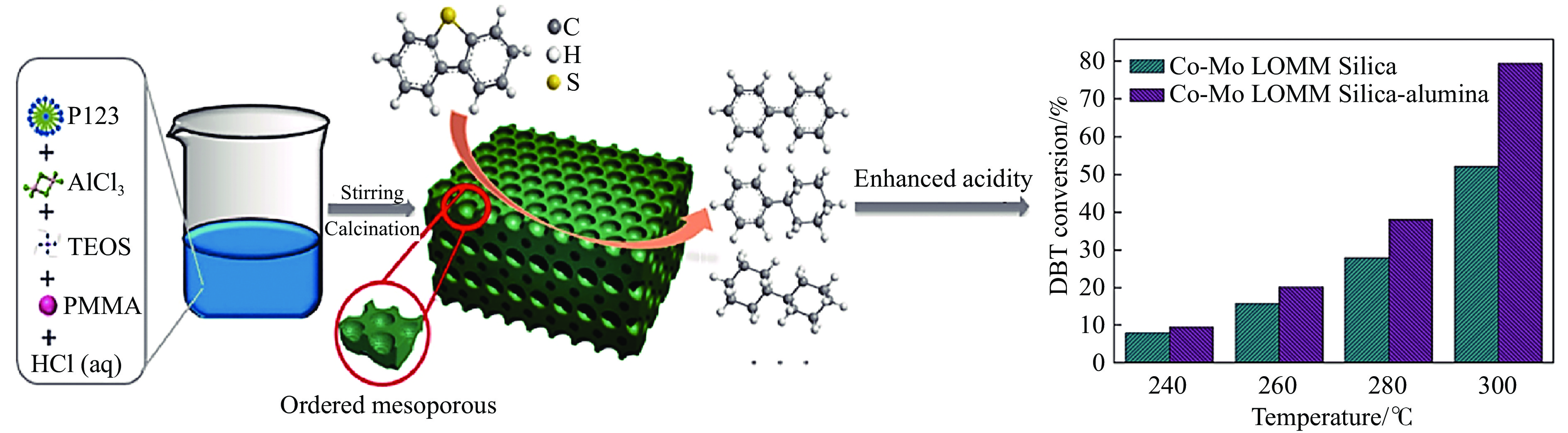
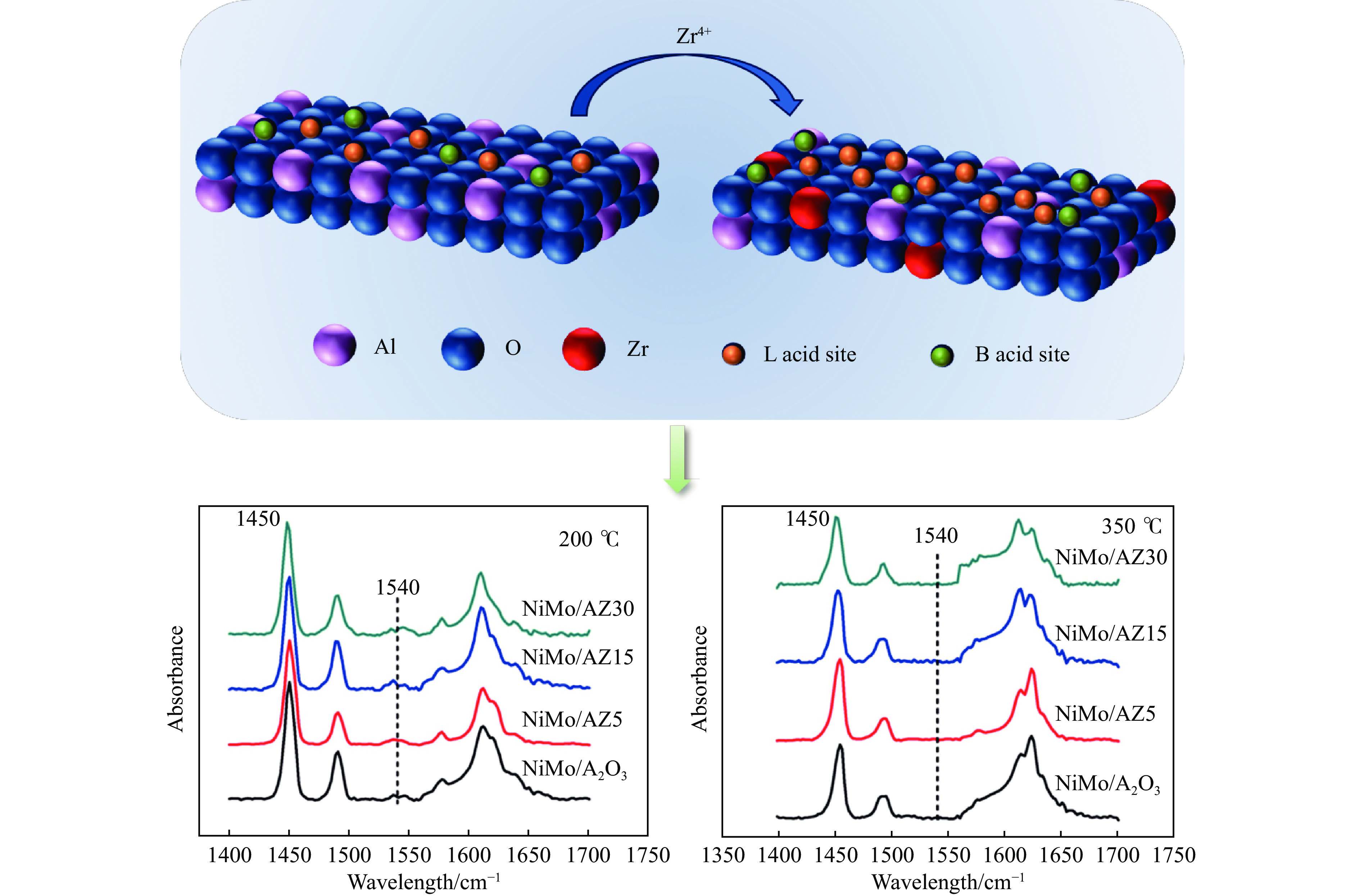
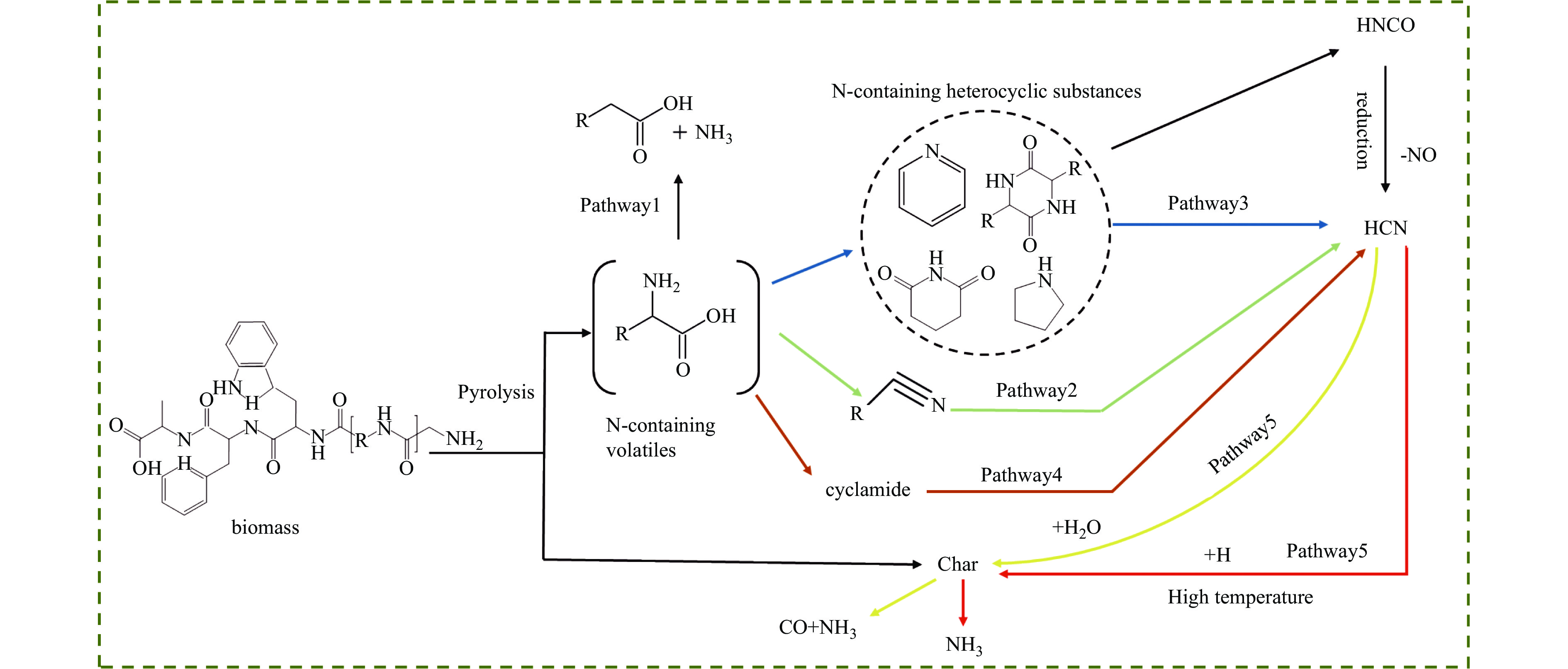
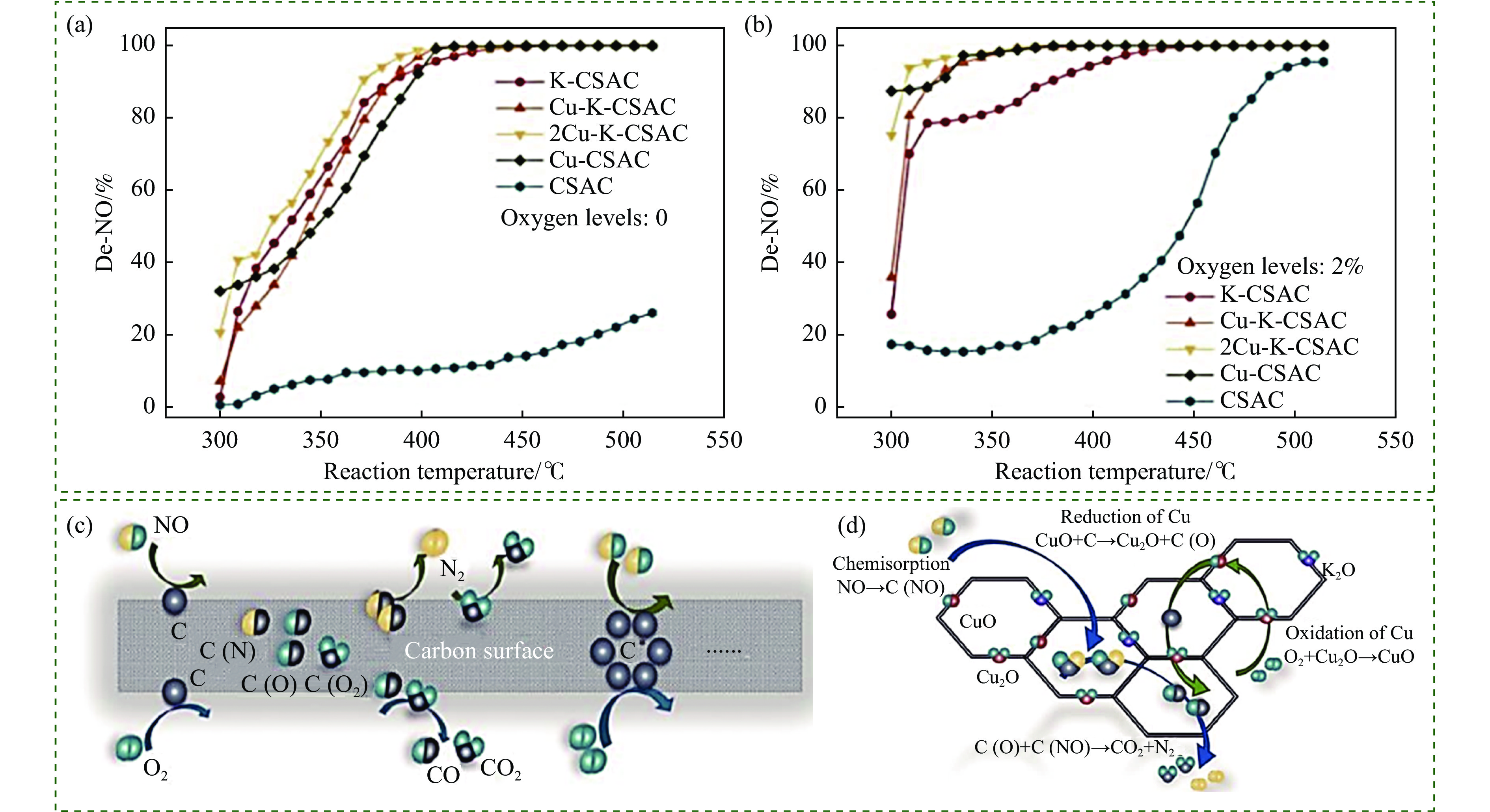
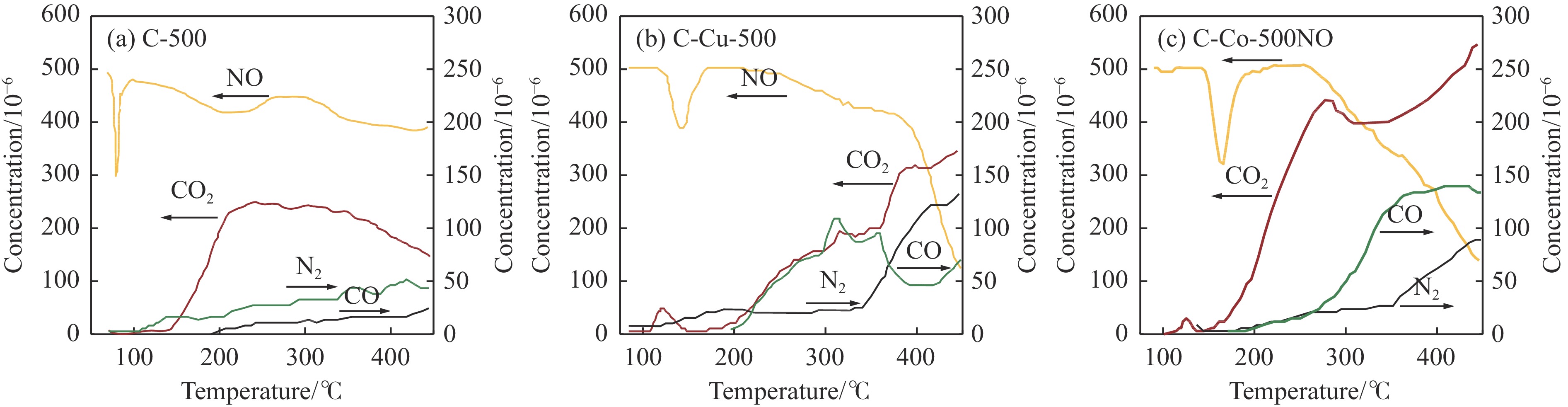
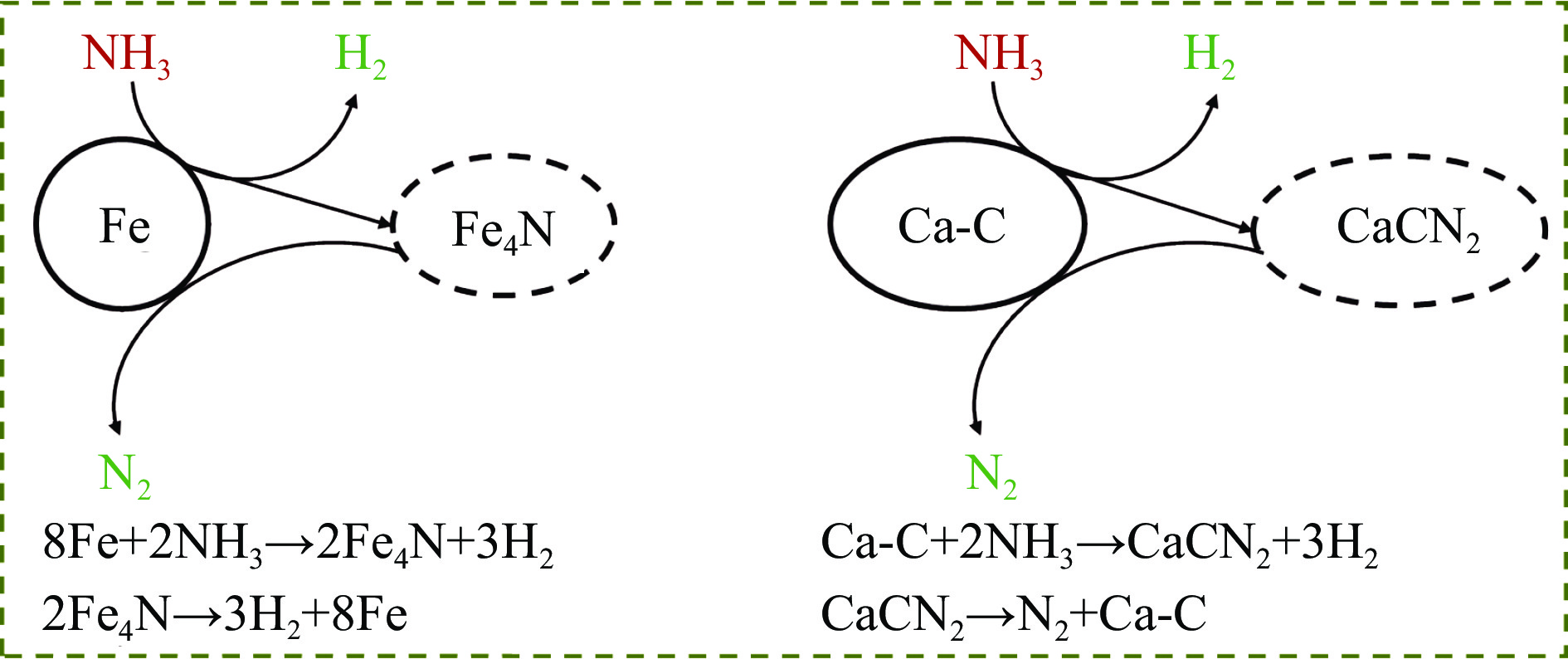
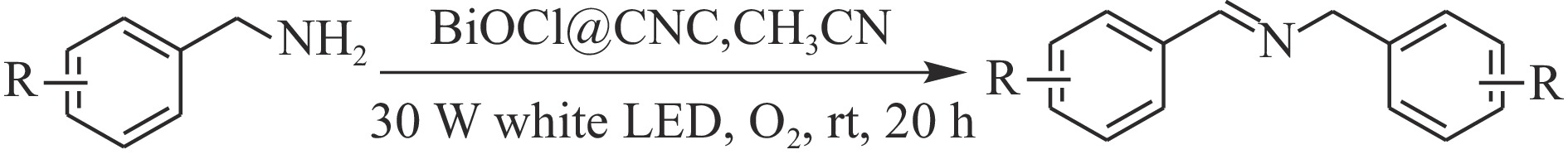Abstract:
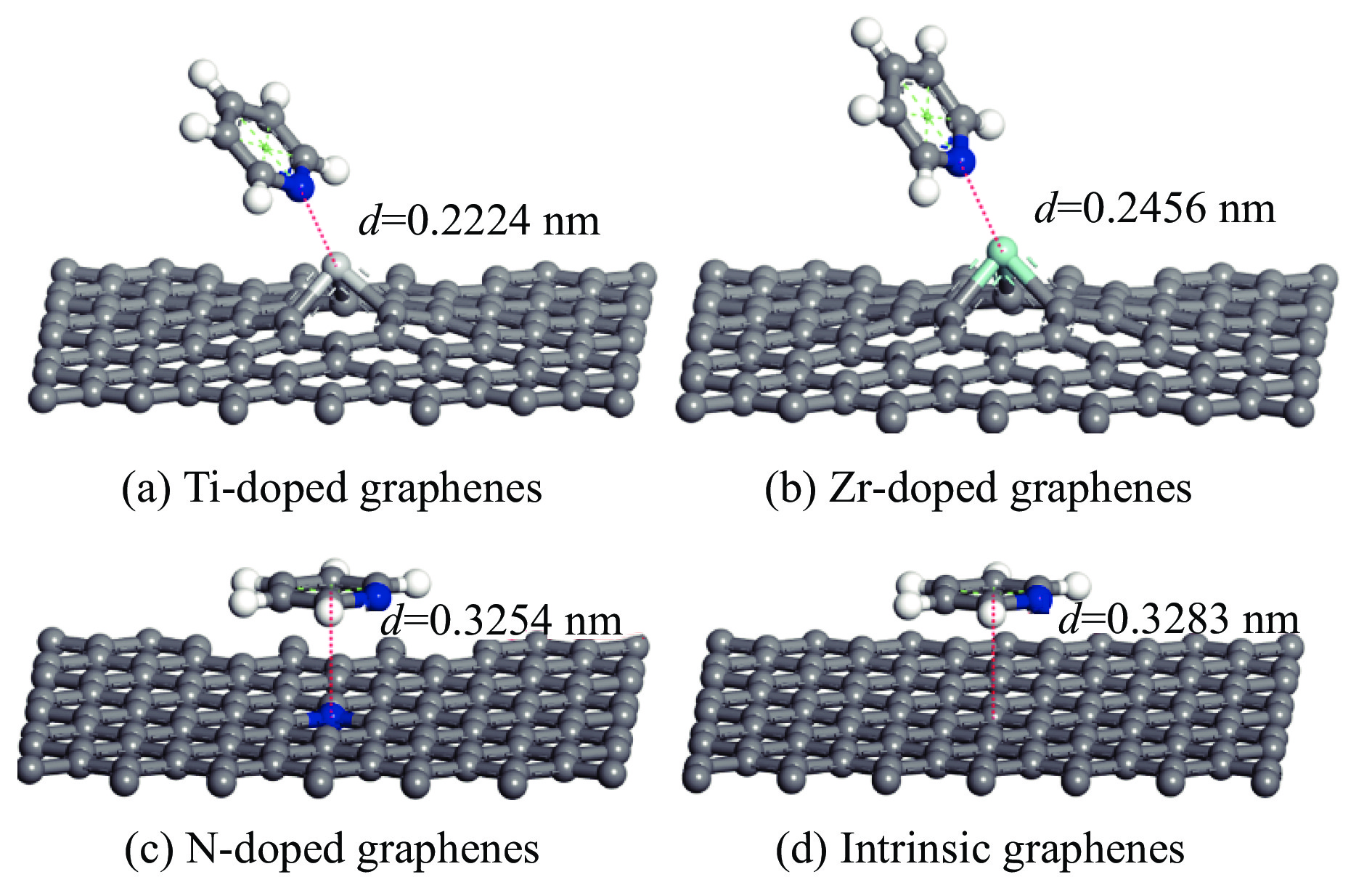
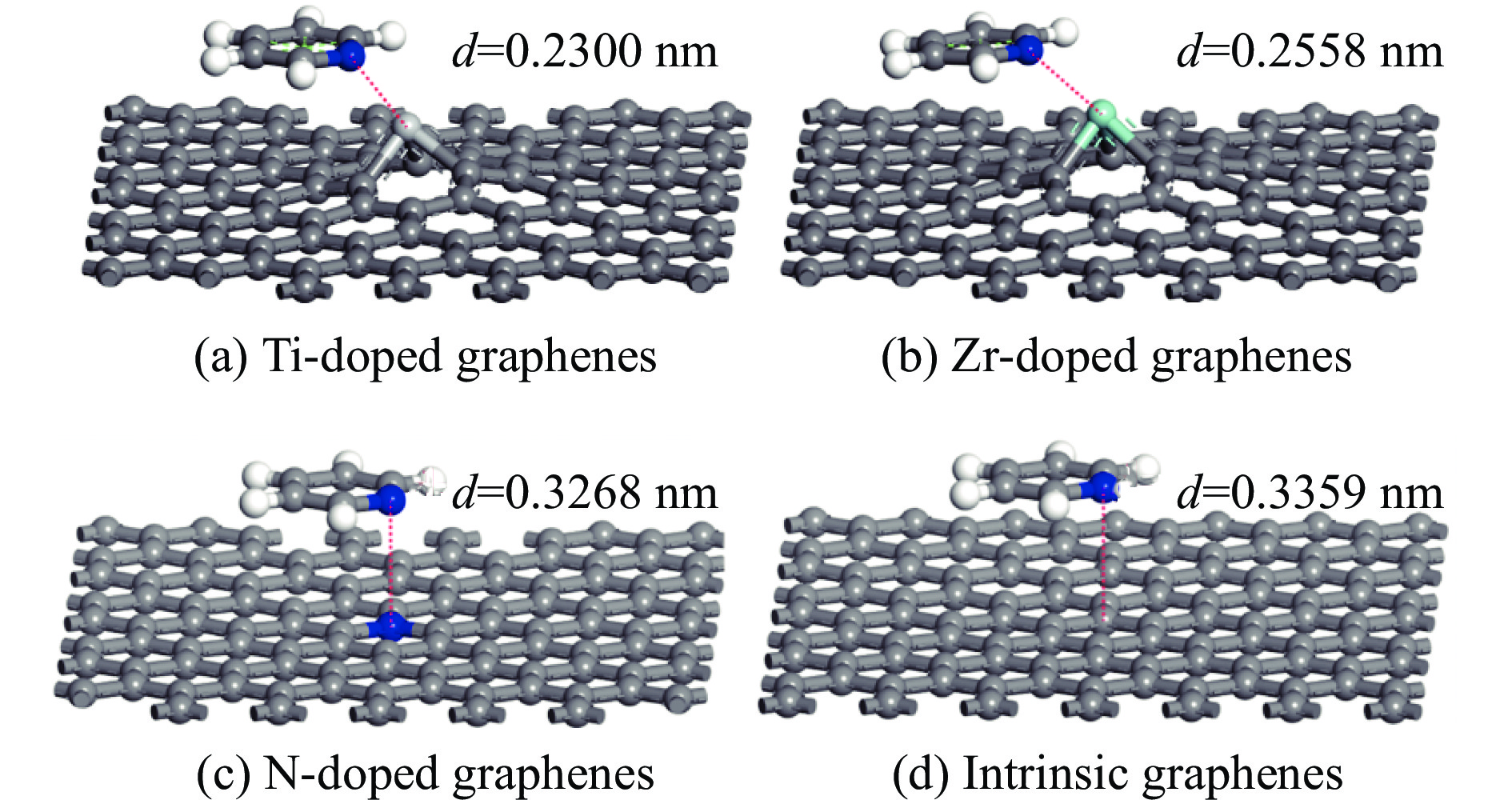
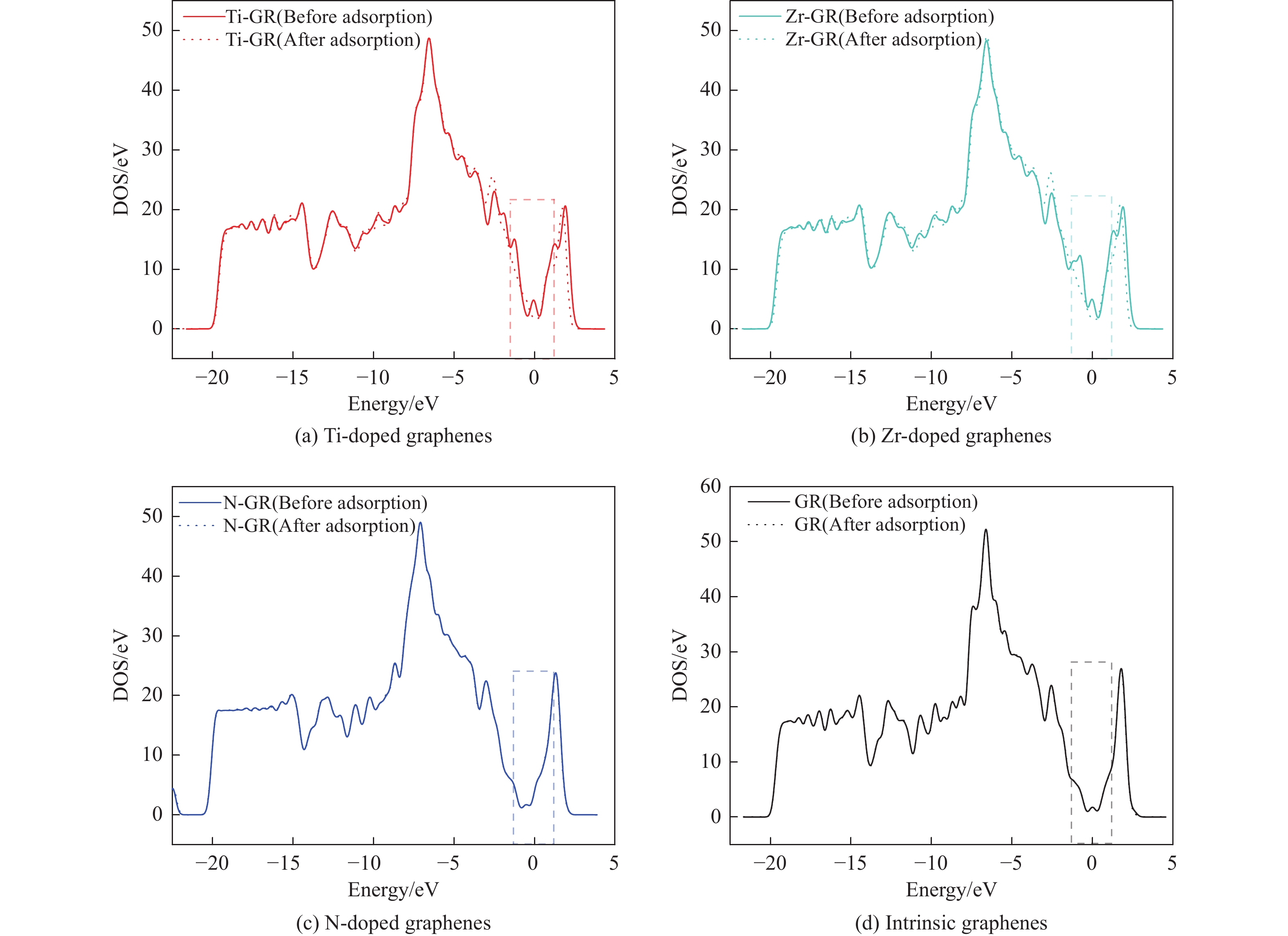
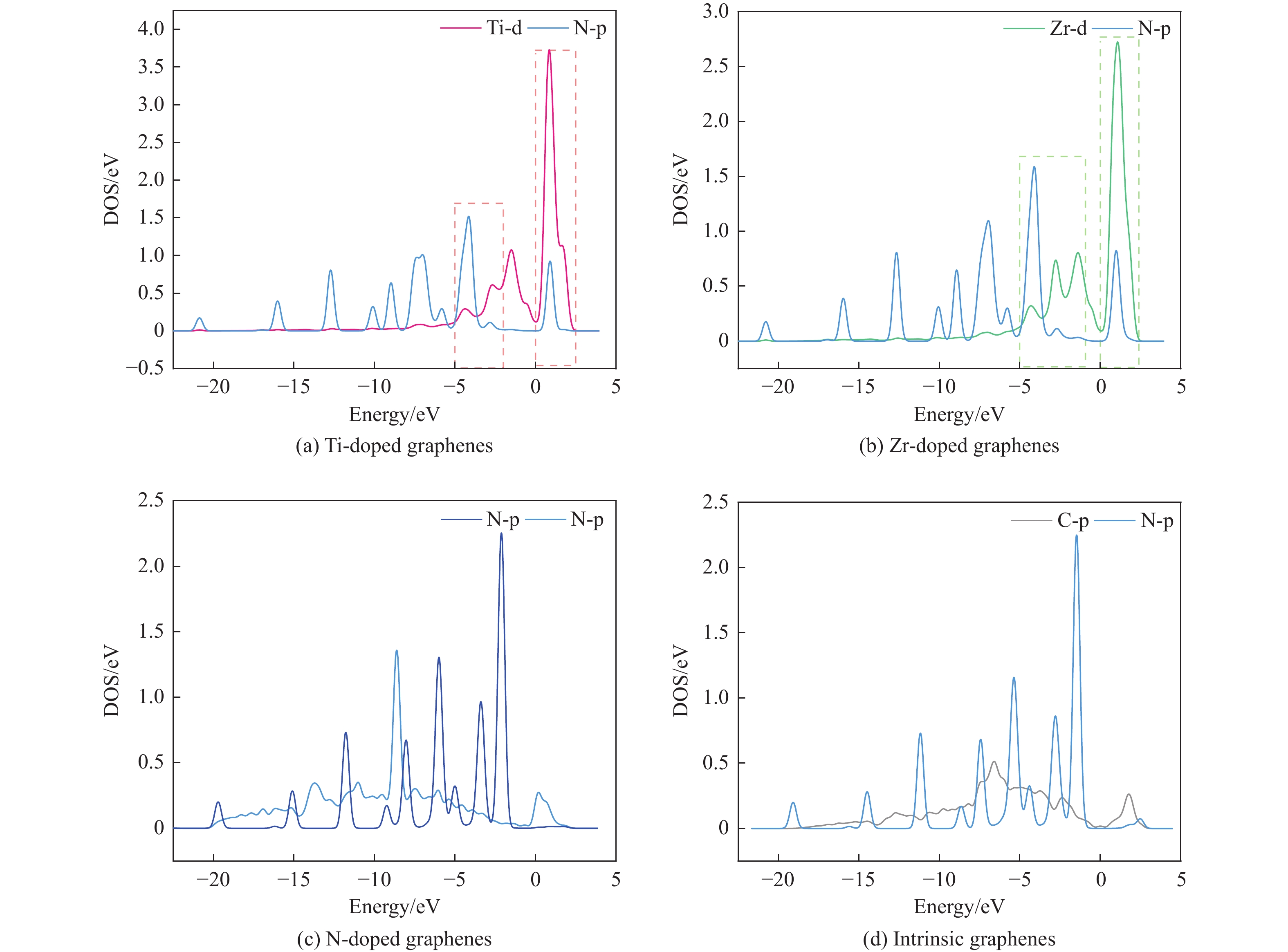
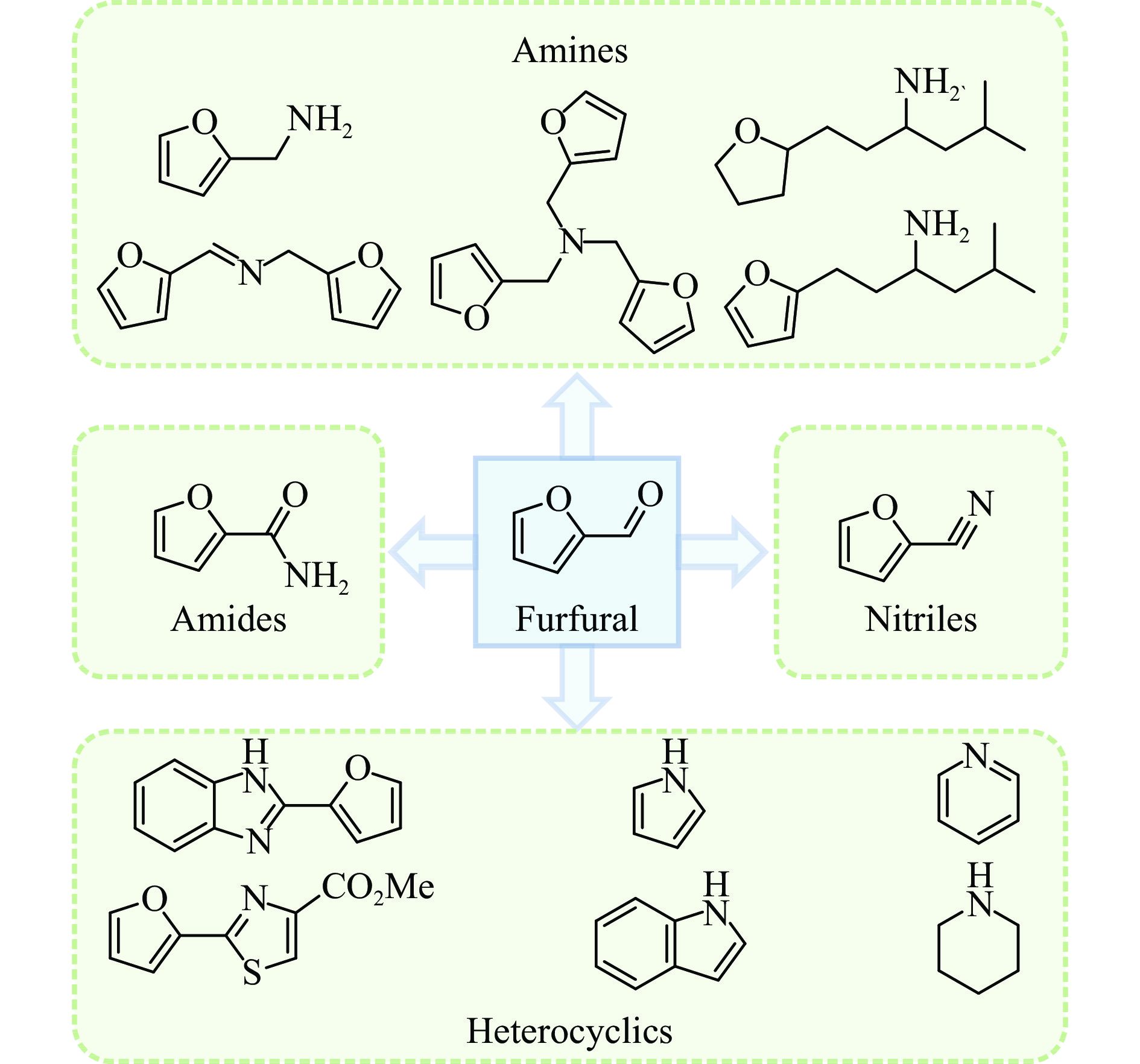
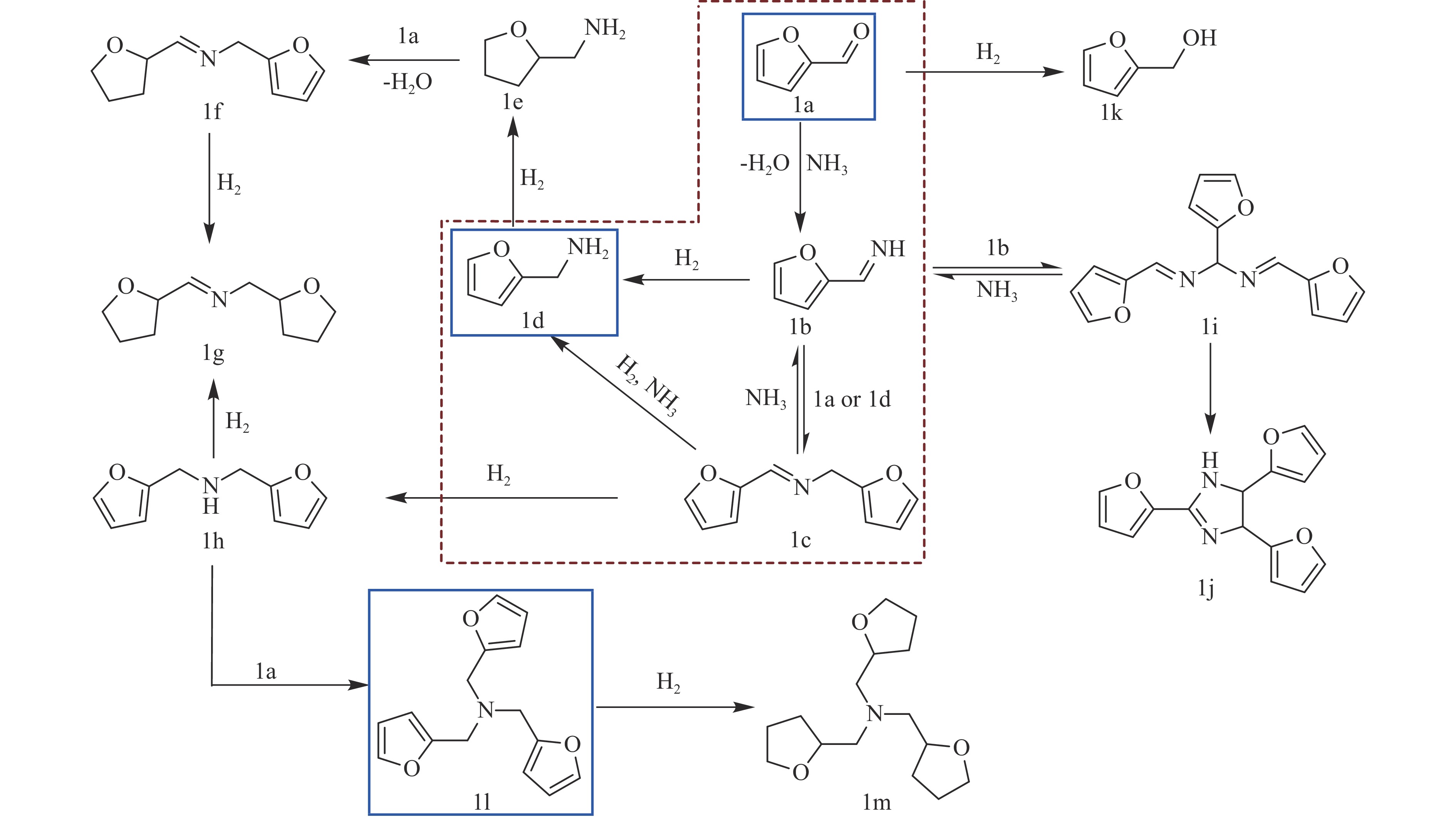
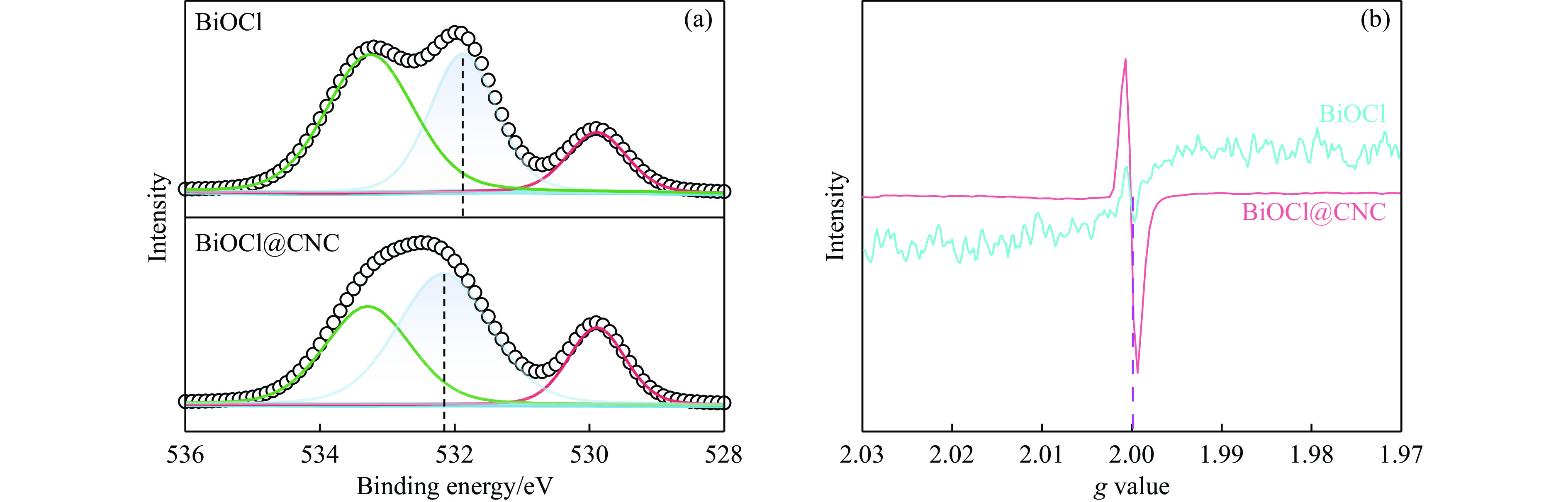
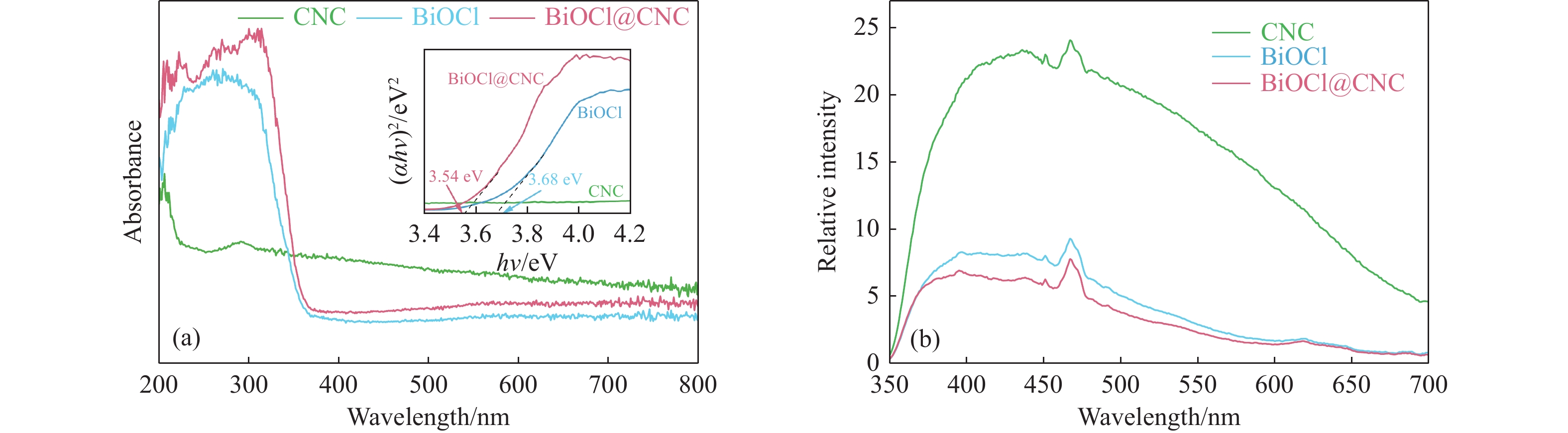
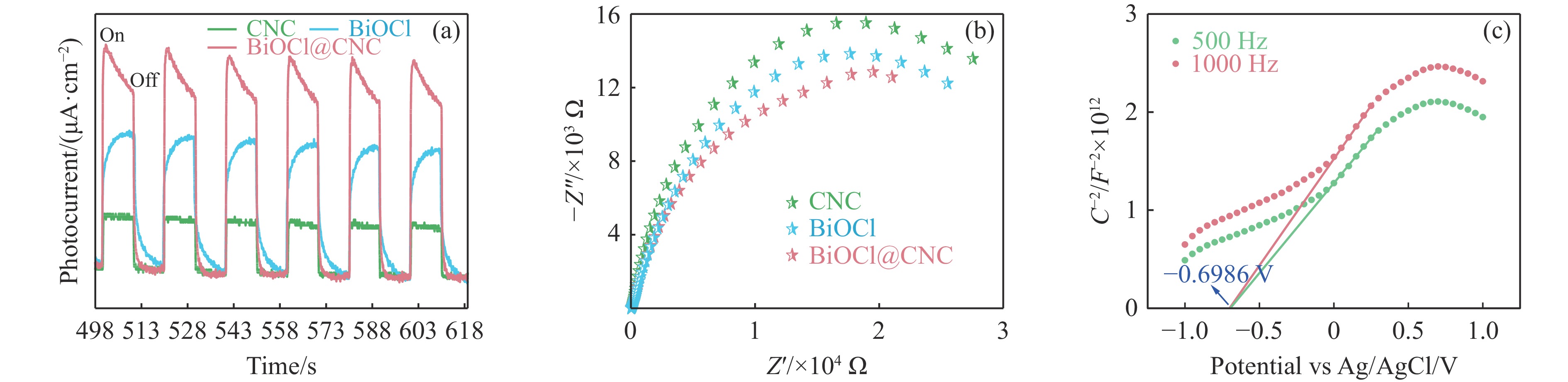
The utilization of biomass holds a great promise to partially replace the non-renewable fossil resources for the production of chemicals and materials for daily use, which could effectively mitigate the challenges associated with global resource scarcity. Furfural, a prominent biomass-derived platform compound derived from the dehydration of xylose in hemicellulose, which is widely used as the key intermediate or solvent in the petrochemicals, coating, pesticides, medicine, synthetic rubber, etc. At the same time, furfural can be converted into a series of high value-added chemicals and fuel, such as alcohols, acids, esters, nitriles, amines, and others due to its active C=O bonds and furan rings. Typical chemical reactions, such as reduction, oxidation, etherification, ammonia oxidation, reduction amination, ring rearrangements and others, are frequently used for the above conversions. Among various chemicals obtained from furfural conversions, nitrogen-containing compounds have attracted considerable attention, owing to the wide applications of such type molecules in the synthesis of drug molecules, bioplastics, and other functional materials. Therefore, using furfural as a raw material to synthesize bio-based nitrogenous compounds represents a cutting-edge research direction. In the presence of nitrogen sources, furfural can be transformed into diverse nitrogen-containing compounds through different reactions, such as reduction amination, ammonia oxidation, oxidative coupling, etc. Varied nitrogen sources (e.g. NH3, N2H4·H2O, NH4HCO2, CH3COONH4, (NH4)2CO3 and others), catalysts, reaction atmospheres, and temperatures can result in distinct target products during furfural conversions. Currently, domestic and foreign research groups have made significant progress on furfural conversions to different nitrogen-containing compounds. Therefore, this review aims to briefly outline the recent achievements in the synthesis of high-value nitrogen-containing compounds from furfural through catalytic conversions over different catalysts. The main content includes: (1) synthesizing amines by reduction aminations, e.g. primary, secondary, and tertiary amines; (2) nitriles production by ammonia oxidation; (3) producing amides by amidation; (4) preparing heterocyclic compounds, such as benzoheterocyclic, thiazole, pyrrole, indole, piperidine and pyridine via oxidative cyclization, decarbonylation-amination, reduction amination, hydrogenation, ring rearrangements. The influences of synthesis methods, catalyst types, reaction pathways, mechanisms, as well as the nitrogen sources, on product distributions were discussed in detail. Considering the pathways and products potentially affected by different nitrogen sources and reaction conditions, future breakthroughs in the synthesis of nitrogen-containing compounds from furfural can be anticipated from the following aspects: (1) By systematically considering the reaction processes and mechanisms, the construction of composite catalysts and precise adjustment of reaction conditions to integrate multiple reaction steps into one is a trend in this research area to attain more efficient and green conversion processes; (2) Combined experimental with theoretical investigations to comprehensively reveal the reaction pathways during the reaction of different nitrogen sources with furfural; (3) Exploration of new chemical conversion routes and catalysts for the production of more novel nitrogen-containing compounds, to further broaden the application areas of furfural based chemicals. In brief, this review provides a systematical review on the production of furfural based nitrogen-containing chemicals, which would benefit the communities working in biomass utilization areas, and also contribute to the establishment of knowledge of the furfural chemical family.